
Issue published November 24, 2025
- Volume 10, Issue 22
- Previous Issue
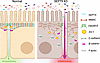
Naydenov et al. report that the septin cytoskeleton localizes to apical junctions of intestinal epithelial cells and maintains barrier integrity, thus protecting the intestinal epithelium from inflammation. The cover image shows the surface of the ileum where SEPT9 is expressed.
On the cover: The septin cytoskeleton is a regulator of intestinal epithelial barrier integrity and mucosal inflammation
-
Research Articles
×
Abstract
Impairment of desmosomal cell-cell adhesion leads to life-threatening diseases, such as the autoimmune skin-blistering disorder pemphigus vulgaris (PV). Disease management strategies that stabilize intercellular adhesion, in addition to the existing immunosuppression therapies, may result in improved clinical outcomes. Previous findings showed that the serine protease inhibitor SERPINB5 promotes intercellular adhesion by binding to and regulating the localization of the desmosomal adapter molecule desmoplakin (DSP) at the plasma membrane. We here show that SERPINB5 overexpression prevents PV-IgG–mediated loss of cell-cell adhesion and DSP dissociation from the cell membrane. We mechanistically demonstrate that SERPINB5 loss deregulates TGF-β signaling, a pathway known to destabilize DSP in keratinocytes. TGF-β signaling was also activated in skin biopsies of patients with PV and keratinocytes treated with PV autoantibodies, suggesting a contribution to disease. Inhibition of TGF-β signaling ameliorated PV-IgG–mediated loss of cell-cell adhesion, increased DSP membrane expression, and prevented PV-IgG–induced blister formation in a human ex vivo skin model. Together, SERPINB5 modulates DSP and intercellular adhesion through the regulation of TGF-β signaling. Further, TGF-β signaling was identified as a potential target for pemphigus treatment.
Authors
Maitreyi Rathod, Mariam Petrosyan, Aude Zimmermann, Maike Märker, Tobias Gosau, Henriette Franz, Tomás Cunha, Dario Didona, Michael Hertl, Enno Schmidt, Volker Spindler
×Abstract
Alzheimer disease (AD) is characterized by plaques and tangles, including calcium dysregulation and glycated products produced by reactive carbonyl compounds. AD brains have increased glyoxalase I (GLO1), a major scavenger of inflammatory carbonyl compounds, at early, but not later, stages of disease. Calcium dysregulation includes calcium leak from phosphorylated ryanodine receptor 2 (pS2808-RyR2), seen in aged macaques and AD mouse models, but the downstream consequences of calcium leak remain unclear. Here, we show that chronic calcium leak is associated with increased GLO1 expression and activity. In macaques, we found age-related increases in GLO1 expression in the prefrontal cortex (PFC), correlating with pS2808-RyR2, and localized to dendrites and astrocytes. To examine the relationship between GLO1 and RyR2, we used S2808D-RyR2 mutant mice exhibiting chronic calcium leak through RyR2, and found increased GLO1 expression and activity in the PFC and hippocampus as early as 1 month and as late as 21 months of age, with a bell-shaped aging curve. These aged S2808D-RyR2 mice demonstrated impaired working memory. As with macaques, GLO1 was expressed in astrocytes and neurons. Proteomics data generated from S2808D-RyR2 synaptosomes confirmed GLO1 upregulation. Altogether, these data suggest potential association between GLO1 and chronic calcium leak, providing resilience in early stages of aging.
Authors
Elizabeth Woo, Dibyadeep Datta, Shveta Bathla, Hannah Beatty, Pinar Caglayan, Ashley Kristant Albizu, TuKiet T. Lam, Jean Kanyo, Mary Kate Joyce, Shannon Leslie, Stacy Uchendu, Jonathan DeLong, Qinyue Stacy Guan, Jiaxin Li, Efrat Abramson, Alison L. Herman, Dawson Cooper, Pawel Licznerski, Tamas L. Horvath, Elizabeth A. Jonas, Angus C. Nairn, Amy F.T. Arnsten, Lauren H. Sansing
×Abstract
Processes that promote white adipocyte inflammatory function remain incompletely defined. Here, we demonstrated that type I interferon–dependent (IFN-I–dependent) skewing of adipocyte glycolysis, nicotinamide adenine dinucleotide (NAD+) utilization, and pyruvate kinase isozyme M2 (PKM2) function may contribute to increased systemic and tissue inflammation and disease severity in obesity. Notably, chemical and/or genetic inhibition of glycolysis, the NAD+ salvage pathway, or PKM2 restricted IFN-I–dependent increase in adipocyte inflammatory cytokine production. Further, genetic or small molecule targeting of PKM2 function in vivo was sufficient to reduce systemic and tissue inflammation and metabolic disease severity in obese mice, in an adipocyte PKM2-dependent manner. Further, white adipose tissue of individuals living with obesity and metabolic disease, compared with metabolically healthy individuals with obesity, showed an increase in expression of inflammatory and metabolic genes, while small molecule targeting of PKM2 function contributed to reduced IFN-I–driven inflammatory cytokine production by primary human adipocytes. Together, our findings invoke the IFN-I/PKM2 axis as a potential target for modulating adipocyte dysregulated inflammation.
Authors
Michelle S.M.A. Damen, Pablo C. Alarcon, Calvin C. Chan, Traci E. Stankiewicz, Hak Chung, Keisuke Sawada, Cassidy J. Ulanowicz, John Eom, Jarren R. Oates, Jennifer L. Wayland, Jessica R. Doll, Rajib Mukherjee, Miki Watanabe-Chailland, Lindsey Romick-Rosendale, Sara Szabo, Michael A. Helmrath, Joan Sanchez-Gurmaches, Maria E. Moreno-Fernandez, Senad Divanovic
×Abstract
Therapeutics blocking PI3K/mTOR complex 1 (mTORC1) are commonly used for tumor treatment, and at times achieve major responses, yet minimal residual disease (MRD) persists, leading to tumor relapse. We developed multiple MRD models both in vitro (rapamycin persistent, RP) and in vivo after mTORC1 inhibition. All 11 RP/MRD cell lines showed complete growth and signaling insensitivity to rapamycin but variable sensitivity to bi-steric mTORC1 inhibitors, with MtorS2035 mutations identified in 4 of 7 RP cell lines. Multiomic analyses identified a pronounced shift toward oxidative phosphorylation and away from glycolysis with increased mitochondrial number in all RP/MRD models. MYC and SWI/SNF expression was significantly enhanced. Both the SWI/SNF inhibitor AU-15330 and the mitochondrial complex I oxidative phosphorylation inhibitor IACS-010759 showed pronounced synergy with bi-steric mTORC1 inhibitors to cause cuproptotic cell death in RP/MRD cells, suggesting these combinations as a potential patient treatment strategy for rapalog resistance.
Authors
Heng Du, Heng-Jia Liu, Magdalena Losko, Yu Chi Yang, Min Yuan, Elizabeth P. Henske, John M. Asara, Mallika Singh, David J. Kwiatkowski
×Abstract
The MTM1 gene encodes myotubularin (MTM1), a phosphatidylinositol 3-phosphate [PI(3)P] lipid phosphatase. Loss-of-function mutations in MTM1 cause X-linked myotubular myopathy (XLMTM), a severe congenital myopathy with no available cure and a poorly understood pathomechanism. The importance of MTM1 enzymatic activity and its PI(3)P substrate in physiology under normal conditions and in XLMTM is unclear. We generated the Mtm1-KI C375S mice in which the endogenous MTM1 was converted to a phosphatase-dead protein. Mutant mice survived a median of 12 weeks and demonstrated progressively impaired motor skills. Observed muscle hypotrophy and reduced force production compared with their WT littermates (~3.9-fold reduction in absolute maximal force) were responsible for these severe phenotypes. A significantly higher level of PI(3)P was found in the muscle of Mtm1-KI C375S mice. Muscle histology and molecular characterization revealed XLMTM hallmarks, with (a) alteration of the mTOR and autophagy pathways correlating with muscle hypotrophy and (b) abnormal myofiber intracellular organization correlating with impaired muscle force. Overall, this study reveals the importance of MTM1 phosphatase activity and related PI(3)P substrate for postnatal muscle maintenance, and it highlights the significance of MTM1 phosphatase activity in the development of X-linked myotubular myopathy.
Authors
Foteini Moschovaki-Filippidou, Christine Kretz, David Reiss, Gaëtan Chicanne, Bernard Payrastre, Jocelyn Laporte
×Abstract
Many patients suffering from inherited diseases do not receive a genetic diagnosis and are therefore excluded as candidates for treatments, such as gene therapies. Analyzing disease gene transcripts from patient cells holds potential for detection and interpretation of causative variants, but may be complicated by unavailability of affected tissue and/or lack of expression of the respective genes in blood or other easily accessible tissues. Using CRISPR/Cas-mediated transcriptional activation (CRISPRa), we developed a robust and efficient approach to activate genes in skin-derived fibroblasts and in freshly isolated peripheral blood mononuclear cells (PBMCs) from healthy individuals. This approach was successfully applied to blood samples from patients with inherited retinal dystrophies (IRDs). We were able to efficiently activate several IRD genes and detect their transcript isoforms using different diagnostically relevant methods such as RT-(q)PCR and long- and short-read RNA sequencing. CRISPRa-mediated transcriptional activation in PBMCs and fibroblasts will contribute to closing the critical gap in the genetic diagnosis of patients with IRD or other inherited diseases.
Authors
Valentin J. Weber, Alice Reschigna, Maximilian J. Gerhardt, Thomas Heigl, Klara S. Hinrichsmeyer, Sander van den Engel, Dina Y. Otify, Zoran Gavrilov, Frank Blaser, Isabelle Meneau, Christian Betz, Hanno J. Bolz, Martin Biel, Stylianos Michalakis, Elvir Becirovic
×Abstract
Atrial fibrillation (AF) is a prevalent arrhythmia with known detriments such as heart failure, stroke, and cognitive decline even in patients without prior stroke. The mechanisms by which AF leads to cognitive dysfunction are yet unknown, and there is a lack of animal models to study this disease process. We previously developed a murine model of spontaneous and prolonged episodes of AF, a double transgenic mouse model with cardiac-specific expression of a gain-of-function mutant voltage-gated sodium channel (DTG-AF mice). Herein, we show, for the first time to our knowledge, a murine model of AF without any cerebral infarcts exhibiting cognitive dysfunction, including impaired visual learning and cognitive flexibility on touch screen testing. Mesenteric resistance arterial function of DTG-AF mice showed significant loss of myogenic tone, increased wall thickness and distensibility, and mitochondrial dysfunction. Brain pial arteries also showed increased wall thickness and mitochondrial enlargement. Furthermore, DTG-AF mice have decreased brain perfusion on laser speckle contrast imaging compared with controls. Cumulatively, these findings demonstrate that AF leads to vascular structural and functional alterations necessary for dynamic cerebral autoregulation, resulting in increased cerebral stress and cognitive dysfunction. Expression of mitochondrial catalase (mCAT) to reduce mitochondrial reactive oxygen species (ROS) was sufficient to prevent vascular dysfunction due to AF, restore perfusion, and improve cognitive flexibility.
Authors
Pavithran Guttipatti, Ruiping Ji, Najla Saadallah, Uma Mahesh R. Avula, Deniz Z. Sonmez, Albert Fang, Eric Li, Amar D. Desai, Samantha Parsons, Parmanand Dasrat, Christine Sison, Yanping Sun, Chris N. Goulbourne, Steven R. Reiken, Elaine Y. Wan
×Abstract
Macular edema (ME) can cause profound vision impairment and occurs in several prevalent retinal diseases, including diabetic retinopathy, choroidal neovascularization, retinal vein occlusion, and uveitis. Retinal edema typically results from dysfunction of the blood-retina barrier (BRB), which is associated with increased retinal expression of complement components. It is unclear whether the classical complement pathway has detrimental or protective roles in the context of BRB dysfunction. Here, we characterized Tspan12-KODBM (disrupted BRB maintenance) mice, a mouse model of cystoid edema generated by genetically and pharmacologically manipulating β-catenin–dependent norrin/frizzled-4 (FZD4) signaling. We assessed BRB function, cystoid edema, electroretinogram, and microglia activation outcomes in an aging study with WT, C1qa-KO, Tspan12-KODBM, and Tspan12-KODBM; C1qa-KO compound mutant mice. Phenotypic analyses and cell-based experiments indicated that C1QA contributes to maintaining basal β-catenin–dependent signaling and that the absence of C1QA exacerbates BRB dysfunction, cystoid edema, and neuroinflammation in Tspan12-KODBM; C1qa-KO compound mutant mice. Activation of β-catenin–dependent signaling by an anti-FZD4 and anti-LRP5 agonistic antibody modality achieved complete resolution of cystoid edema. This study shows that reducing or enhancing norrin/FZD4 signaling can increase or decrease cystoid edema, respectively, underscoring its potential as a therapeutic target in ME. Furthermore, this study provides insights into the contribution of C1QA to BRB maintenance.
Authors
Lingling Zhang, Jacklyn Levey, Md. Abedin, Ha-Neul Jo, Emmanuel Odame, Miranda Howe, Kaia L. Douglas, Elise Thoreen, Scott W. McPherson, Heidi Roehrich, Somasekar Seshagiri, Stephane Angers, Zhe Chen, Harald J. Junge
×Abstract
Fibroblast to myofibroblast transition is a critical event required for effective tissue repair. In pathologic wound repair processes, such as type 2 diabetes (T2D), fibroblast to myofibroblast transition is impaired. The exact factors that control this transition in wounds are unclear. Here, using human tissue and murine transgenic models, we show that the histone methyltransferase SETDB2 is elevated in diabetic wound fibroblasts and TNF-α represses fibroblast to myofibroblast transition via Setdb2. We identified that TNF-α increases Setdb2 in fibroblasts via a JAK1,3/STAT3 signaling pathway, where pharmacologic or genetic manipulation of this pathway altered Setdb2 in fibroblasts. We also found that fibroblasts treated with pro-inflammatory macrophage supernatants displayed increased Setdb2 and downregulated myofibroblast genes; inhibition of the TNF-α receptor reduced the upregulation of Setdb2. In diabetes, we showed that TNF-α signaling was increased in wound fibroblasts, which functions to increase Setdb2 expression and represses fibroblast to myofibroblast transition. Fibroblast-specific knockdown of SETDB2 and therapeutic inhibition of JAK1,3/STAT3 improved diabetic wound repair, where wound fibroblasts expressed increased myofibroblast genes. This study is the first to our knowledge to identify an epigenetic mechanism for reduced fibroblast to myofibroblast transition in diabetic wounds. Therapeutic targeting of the TNF-α/STAT3/SETDB2 axis in wound fibroblasts may improve diabetic wound healing.
Authors
Tyler M. Bauer, Kevin D. Mangum, Samuel D. Buckley, James Shadiow, Amrita D. Joshi, Christopher O. Audu, Jadie Y. Moon, Lindsey D. Hughes, Rachel Bogel, Lam C. Tsoi, Qinmennge Li, He Zhang, Steven Kunkel, Johann E. Gudjonsson, Frank M. Davis, Katherine A. Gallagher
×Abstract
Intestinal epithelial barrier integrity is essential for human health, and its disruption induces and exacerbates intestinal inflammatory disorders. While the epithelial cytoskeleton is critical for maintaining gut barrier-integrity, the role of septins — a family of GTP-binding, cytoskeletal proteins — is largely unknown. This highlights an important knowledge gap, as dysfunction of septins, and specifically septin 9 (SEPT9), is associated with intestinal pathologies. We determined that SEPT9 localizes to the apical junctions of intestinal epithelial cells (IECs), overlapping with both tight and adherens junctions. IEC-specific ablation of SEPT9 in mice resulted in leaky gut, due to mislocalization of junctional proteins, and increased susceptibility to experimental colitis. Consistently, SEPT9 expression was significantly reduced in intestinal mucosa of patients with inflammatory bowel disease (IBD). Using affinity-purification mass spectrometry, super-resolution imaging, and genetic KO, we determined that SEPT9 interacts with and is necessary to recruit nonmuscle myosin IIC (NMIIC) to the IEC perijunctional actomyosin belt. Loss of NMIIC also caused IEC barrier disruption. In summary, SEPT9 regulates intestinal barrier integrity by supporting the assembly of tight and adherens junctions through NMIIC recruitment to the actomyosin belt. The septin cytoskeleton safeguards the intestinal mucosa during acute inflammation, and its disruption in IBD suggests a loss of this protective function.
Authors
Nayden G. Naydenov, Gaizun Hu, Dominik Robak, Atif Zafar, Khosiyat Makhmudova, Susana Lechuga, Yuta Ohno, Naseer Sangwan, Saikat Bandyopadhyay, Ryan Musich, Erin Jeffery, Lei Sun, Armando Marino-Melendez, Florian Rieder, Gloria Sheynkman, Andrei I. Ivanov, Seham Ebrahim
×Abstract
Genetic diseases such as ion channelopathies substantially burden human health. Existing treatments are limited and not genotype specific. Here, we report a 2-step high-throughput approach to rapidly identify drug candidates for repurposing as genotype-specific therapy. We first screened 1,680 medicines using a thallium-flux trafficking assay against Kv11.1 gene variants causing long QT syndrome (LQTS), an ion channelopathy associated with fatal cardiac arrhythmia. We identified evacetrapib as a suitable drug candidate that improves membrane trafficking and activates channels. We then used deep mutational scanning to prospectively identify all Kv11.1 missense variants in an LQTS hotspot region responsive to treatment with evacetrapib. Combining high-throughput drug screens with deep mutational scanning establishes a paradigm for mutation-specific drug discovery translatable to personalized treatment of carriers with rare genetic disorders.
Authors
Christian L. Egly, Alex Shen, Tri Q. Do, Carlos Tellet Cabiya, Paxton A. Ritschel, Suah Woo, Matthew Ku, Brian P. Delisle, Brett M. Kroncke, Björn C. Knollmann
×Abstract
Peripheral helper T (Tph) and follicular helper T (Tfh) cells are key regulators of B cell differentiation and antibody production, making them promising targets for autoimmune disease treatment. However, their differentiation mechanisms differ significantly between humans and mice, limiting drug validation in mouse models. Here, we present a simple and effective method for in vivo proliferation of human Tph/Tfh and B cells. We discovered that after depleting CD8+ T cells of human peripheral blood mononuclear cell–transferred immunodeficient mice (CD8TΔhPBMC mice), human Tph/Tfh cells and B cells proliferated markedly in the spleen compared with those in human PBMC–transferred immunodeficient mice (hPBMC mice). Transcriptome analysis confirmed proliferating cells’ close resemblance to human Tph/Tfh cells. Furthermore, multicolor flow cytometry revealed CXCL13+ Tph cells infiltrating Sjögren’s syndrome–associated (SjS-associated) organs, such as salivary glands. Single-cell RNA sequencing identified IL-21+CXCL13+IFN-γ+ICOS+TIGIT+GPR56+ Tph cells in the salivary glands. These findings are consistent with reduced saliva volume and elevated SjS markers, such as anti-SSA antibody, in these mice, which were both ameliorated by immunosuppressants. In vitro, CD8+ T cells from hPBMC mice induced B cell apoptosis and inhibited Tph/Tfh differentiation. This model advances understanding of human Tph/Tfh cell biology and offers a valuable platform for studying SjS and therapeutic targets.
Authors
Mariam Piruzyan, Sota Fujimori, Ryota Sato, Yuki Imura, Sachiko Mochiduki, Kana Takemoto, Akiko Nishidate, Yuzo Koda
×Abstract
Poor skeletal muscle fitness contributes to many chronic disease states, including obesity, heart failure, primary muscle disorders, and age-related sarcopenia. Receptor-interacting protein 140 (RIP140) is a striated muscle–enriched nuclear receptor coregulator known to suppress mitochondrial oxidative capacity. To investigate the role of RIP140 in skeletal muscle, striated muscle–specific RIP140-deficient (strNrip1–/–) mice were generated and characterized. strNrip1–/– mice displayed an enhanced endurance performance phenotype. RNA-sequence (RNA-seq) analysis of glycolytic fast-twitch muscle from strNrip1–/– mice identified a broad array of differentially upregulated metabolic and structural muscle genes known to be induced by endurance training, including pathways involved in mitochondrial biogenesis and respiration, fatty acid oxidation, slow muscle fiber type, and angiogenesis. In addition, muscle RIP140 deficiency induced expansive neuromuscular junction (NMJ) remodeling. Integration of RNA-seq results with CUT&RUN analysis of strNrip1–/– myotubes identified Wnt16 as a candidate effector for the NMJ biogenesis in RIP140-deficient skeletal myotubes. We conclude that RIP140 serves as a physiological “rheostat” for a broad coordinated network of metabolic and structural genes involved in skeletal muscle fitness.
Authors
Elizabeth Pruzinsky, Kirill Batmanov, Denis M. Medeiros, Sarah M. Sulon, Brian P. Sullivan, Tomoya Sakamoto, Teresa C. Leone, Tejvir S. Khurana, Daniel P. Kelly
×Abstract
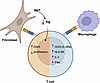
In the rheumatoid arthritis (RA) synovium, resident fibroblast-like synoviocytes (FLS) express MHC class II molecules (HLA-D) but lack the costimulatory signals typically required for T cell activation. Here, we demonstrate that antigen presentation by FLS induces a distinct T cell activation state characterized by high CD69 yet reduced CD25 and HLA-DR expression, suppressed proliferation, and decreased effector cytokine production compared with professional antigen-presenting cells (APCs), such as macrophages. FLS were also capable of suppressing macrophage-induced T cell activation, underscoring their dominant immunomodulatory role in the synovial microenvironment. Mechanistically, we identify indoleamine 2,3-dioxygenase–mediated (IDO1-mediated) tryptophan depletion as the primary driver of FLS-induced T cell hyporesponsiveness. Spatial transcriptomics revealed colocalization of IDO1 and CD69 within ectopic lymphoid structures in RA synovium, further supporting the in vivo relevance of this pathway. These findings provide the groundwork for positioning FLS as critical T cell regulators in RA and highlight the importance of preserving their immunosuppressive properties when therapeutically targeting pathogenic FLS functions.
Authors
Melissa R. Romoff, Preethi K. Periyakoil, Edward F. DiCarlo, Daniel Ramirez, Susan M. Goodman, Christina S. Leslie, Alexander Y. Rudensky, Laura T. Donlin, Melanie H. Smith
×A multiomics analysis identifies retinol metabolism in fibroblasts as a key pathway in wound healing
Abstract
Impaired wound healing poses a major and increasingly frequent health problem. Among the key players in the healing process are fibroblasts, but their metabolic profile in healing wounds is largely unknown. Using a combination of transcriptomics, targeted proteomics, and metabolomics, we identified retinol metabolism as a top regulated pathway in wound fibroblasts. This is functionally relevant, since even a mild retinol deficiency caused a delay in wound closure and reepithelialization, which mainly resulted from misdirected keratinocyte migration on the new granulation tissue. Quantitative proteomics identified integrin subunit α11 as a less abundant protein in wounds of mice subjected to a retinol-deficient diet. Reduced levels of this fibroblast-specific protein likely altered the granulation tissue matrix, which in turn affected reepithelialization. These results provide a comprehensive overview of the transcriptome, proteome, and metabolome of wound fibroblasts and identify retinol metabolism in fibroblasts as a key regulator of tissue repair.
Authors
Till Wüstemann, Elizabeta Madzharova, Mateusz S. Wietecha, Norbert B. Ghyselinck, Marcus Höring, Gerhard Liebisch, Nicola Zamboni, Ulrich auf dem Keller, Sabine Werner
×Abstract
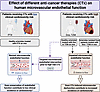
BACKGROUND Cardiotoxicity is a major complication of anticancer therapy (CTx); however, the effect of CTx on the microcirculation is not well defined. This study evaluated the effect of CTx on microvascular function in patients with breast cancer (PwBC).METHODS Endothelial function and angiogenic potential were assessed in arterioles and adipose biopsies obtained from PwBC undergoing CTx (longitudinal and cross-sectional) and in healthy arterioles exposed to doxorubicin (Dox), trastuzumab (TZM), or paclitaxel (PTX) ex vivo. VEGF-B protein was used to test feasibility of targeted intervention.RESULTS PwBC treated with Dox and/or TZM developed profound microvascular endothelial dysfunction that persisted for ≥ 9 months after treatment cessation. Angiogenic potential was reduced during CTx and recovered within 1 month. Gene expression related to angiogenesis and inflammation changed over the course of clinical treatment. Adipose arterioles from healthy donors developed endothelial dysfunction when exposed to Dox or TZM ex vivo. PTX, which poses minimal cardiovascular risk, had no effect on vasomotor function. Ex vivo exposure to Dox or PTX suppressed angiogenic potential, whereas TZM had no effect. VEGF-B protein preserved endothelial function in arterioles exposed to Dox or TZM ex vivo.CONCLUSION PwBC undergoing treatment with Dox and/or TZM develop prolonged microvascular endothelial dysfunction that is recapitulated in healthy arterioles exposed to Dox or TZM ex vivo. Targeted intervention with VEGF-B protects against direct Dox- or TZM-induced vascular toxicity in human arterioles ex vivo.FUNDING NIH, American Heart Association, WeCare Foundation, Medical College of Wisconsin, Advancing a Healthier Wisconsin, Jenny and Antti Wihuri Foundation.
Authors
Janée D. Terwoord, Laura E. Norwood Toro, Shelby N. Hader, Stephen T. Hammond, Joseph C. Hockenberry, Jasmine Linn, Ibrahim Y. Vazirabad, Amanda L. Kong, Alison J. Kriegel, Ziqing Liu, Riikka M. Kivelä, Gillian Murtagh, David D. Gutterman, Andreas M. Beyer
×Abstract
Laminin-α2–related congenital muscular dystrophy (LAMA2-CMD) is a severe neuromuscular disorder caused by mutations in the LAMA2 gene, leading to loss of heterotrimers laminin-211/221, key components of the skeletal muscle extracellular matrix. Their absence disrupts adhesion between the cytoskeleton and extracellular matrix, resulting in progressive muscle wasting. Laminin-211/221 interacts with adhesion complexes such as the dystrophin/utrophin glycoprotein complex and α7β1-integrin. However, the regulatory mechanisms of these laminin-binding complexes and the broader role of laminin’s influence on the formation of the macromolecular network in skeletal muscle remain unclear. We previously demonstrated that delivering mouse laminin-111 to the dyW–/– mouse model of LAMA2-CMD prevented disease progression, improved strength, and extended survival. We hypothesize that laminin-111, the embryonic laminin isoform, restores key adhesion-signaling networks. Using spatial proteomics on patient and mouse muscle, we identified loss of essential signaling components: heat shock proteins 27 and 70, c-Jun N-terminal kinase, and glucose transporter 1 in laminin-α2–deficient muscle. Treatment with recombinant human laminin-111 (rhLAM-111) restored protein localization, reduced ROS, and promoted glycolytic, prosurvival signaling. These findings highlight laminin’s role in maintaining muscle homeostasis and metabolism and support the therapeutic potential of rhLAM-111 for treating LAMA2-CMD by restoring adhesion and intracellular signaling in dystrophic muscle.
Authors
Hailey J. Hermann, Ryan D. Wuebbles, Marisela Dagda, Axel Muñoz, Lauren L. Parker, Paula C. Guzman, Lola T. Byrne, Steven A. Moore, Dean J. Burkin
×Abstract
IDH1/2 mutations (IDHmut) increase methylation of DNA and histones in gliomas. IDHmut inhibitors are effective against low-grade IDHmut gliomas, but new strategies against high-grade IDHmut gliomas are needed. Although histone deacetylase inhibitors (HDACi) are ineffective against IDHwt glioblastoma (GBM), their potential in IDHmut gliomas has not been extensively studied. We previously established that IDHmut gliomas are more sensitive to HDACi than IDHwt GBM. Here we show that IDHmut is associated with greater sensitivity to HDACi only in glioma, not in IDHmut chondrosarcoma or cholangiocarcinoma. While HDACi induced more histone acetylation and gene regulation in IDHmut glioma than in IDHwt GBM, such acetylation was mostly within gene deserts, whereas IDHmut glioma promoters paradoxically lost histone acetylation. Two mediators of HDACi resistance, YAP and TAZ, were methylated and suppressed in IDHmut gliomas but not in other IDHmut cancers. Inducing YAP or TAZ expression in IDHmut gliomas conferred resistance to HDACi. Finally, belinostat extended in vivo survival only in IDHmut glioma models, not in IDHmut GBM models. Our findings provide a mechanistic rationale for further studies of HDACi in patients with IDHmut glioma, as well as the potential use of YAP/TAZ as a biomarker of HDACi sensitivity in cancers.
Authors
Thomas K. Sears, Matthew McCord, Wenxia Wang, Alicia Steffens, Kathleen McCortney, Rahul Chaliparambil, Jann N. Sarkaria, Craig M. Horbinski
×Abstract
HOXB13 is a prostate-specific transcription factor best known for its role as an androgen receptor (AR) cofactor. Recent evidence suggests that HOXB13 plays critical AR-independent functions in repressing lipogenic programs and promoting prostate cancer (PCa) metastasis. However, the mechanisms linking HOXB13 loss to tumor metastasis remain unclear. Here, we show that p300 and CBP co-occupy lipogenic enhancers suppressed by HOXB13 and HDAC3 and are essential for enhancer activation and target gene expression following HOXB13 depletion. Loss of HOXB13 induces lipid-sensitive matrix metalloproteinases (MMPs), promoting increased cell motility. Importantly, pharmacological inhibition of p300 and CBP blocks HOXB13-loss-driven lipogenesis, reduces MMP expression, and decreases cell migration in vitro and tumor metastasis in vivo. Analysis of clinical samples revealed that HOXB13 expression is reduced in metastatic hormone-sensitive PCa compared with matched primary tumors, further supporting its role in tumor metastasis. These findings demonstrate that HOXB13 downregulation promotes PCa metastasis through p300- and CBP-dependent lipogenic and motility pathways, which may be targeted by p300 inhibition.
Authors
Xiaodong Lu, Liu Peng, Qi Chu, Samantha Ye, Mingyang Liu, Maha Hussain, Mehmet A. Bilen, Lara R. Harik, Jonathan Melamed, Jonathan C. Zhao, Jindan Yu
×Abstract
Empirical data from survivors of Lassa fever and experimental disease modeling efforts, particularly those using mouse models, are at odds with respect to T cell–mediated pathogenesis. In mice, T cells have been shown to be imperative in disease progression and lethality, whereas in humans, an early and robust T cell response has been associated with survival. Here, we assessed the role of CD4+ and CD8+ T cells on disease progression and severity of Lassa virus infection in a nonhuman primate model. Using an antibody-mediated T cell depletion strategy prior to and after inoculation, we were able to examine Lassa virus infection in the absence of specific T cell responses. In animals depleted for either CD4+ or CD8+ T cells, Lassa virus infection remained uniformly lethal, with only a slight delay in disease progression was observed in the CD4-depleted group when compared with nondepleted controls. Milder pulmonary pathology was noticed in the absence of CD4+ or CD8+ T cells. Overall, our findings suggest that T cells have a limited effect on the development of Lassa fever in nonhuman primates.
Authors
Jérémie Prévost, Nikesh Tailor, Geoff Soule, Jonathan Audet, Yvon Deschambault, Robert Vendramelli, Jessica Prado-Smith, Kevin Tierney, Kimberly Azaransky, Darwyn Kobasa, Chad S. Clancy, Heinz Feldmann, Kyle Rosenke, David Safronetz
×Abstract
Long-chain fatty acid oxidation disorders (LC-FAODs) cause energy deficits in heart and skeletal muscle that are only partially corrected by current medium-chain lipid therapies such as triheptanoin. We find that heart and muscle lack medium-chain acyl-CoA synthetases, limiting the capacity for β-oxidation of medium-chain fatty acids. Instead, heart and muscle mitochondria robustly respire on medium-chain acylcarnitines. The mitochondrial matrix enzyme carnitine acetyltransferase (CrAT) efficiently converts orally delivered octanoylcarnitine (C8-carnitine) to octanoyl-CoA for energy generation. C8-carnitine exhibits twice the oral bioavailability of triheptanoin and distributes to muscle and heart. A single oral dose significantly enhances grip strength and treadmill endurance while attenuating lactic acidosis in 2 mouse models of LC-FAODs. Thus, medium-chain acylcarnitines overcome a previously unrecognized metabolic bottleneck in LC-FAOD muscle and may represent an alternative to triglyceride-based therapies for bioenergetic disorders.
Authors
Keaton J. Solo, Yuxun Zhang, Sivakama S. Bharathi, Bob B. Zhang, Adam C. Richert, Alexandra V. Schmidt, Clinton Van’t Land, Olivia D’Annibale, Timothy C. Wood, Eric S. Goetzman








In-Press Preview - More
Abstract
Autoimmune diabetes encompasses rapidly progressive type 1 diabetes mellitus (T1D) and indolent latent autoimmune diabetes in adults (LADA), representing distinct inflammatory set points along a shared autoimmune spectrum. Yet the immunological mechanisms that determine these divergent inflammatory states remain unresolved. We performed single-cell RNA sequencing with paired T and B cell receptor profiling on over 400,000 peripheral blood mononuclear cells (PBMCs) from patients with LADA, newly diagnosed T1D, and healthy controls. PBMC composition was comparable across cohorts, indicating that qualitative rather than quantitative immune differences underlie disease heterogeneity. In T1D, pan-lineage activation of NF-κB, EGFR, MAPK, and hypoxia pathways, coupled with a TNF-centered communication hub, enhanced MHC signaling, and disrupted adhesion, promoted systemic inflammation. LADA, by contrast, exhibited global suppression of NF-κB/EGFR activity, retention of moderate JAK/STAT tone, reinforced natural killer cell inhibitory checkpoints via HLA-C–KIR2DL3/3DL1 interaction, and stabilized CD8⁺ T cell synapses through HLA-C–CD8 binding, collectively restraining effector activation. Single-cell V(D)J analysis revealed multiclonal, patient-unique adaptive repertoires, emphasizing the primacy of signaling context over receptor convergence. These findings define autoimmune diabetes as an inflammatory–inhibitory set-point continuum, positioning the NF-κB/EGFR–JAK/STAT gradient and HLA-C–KIR axis as potential therapeutic targets to preserve residual β-cell function.
Authors
Ivan I. Golodnikov, Elizaveta S. Podshivalova, Vadim I. Chechekhin, Anatoliy V. Zubritskiy, Alina A. Matrosova, Nikita A. Sergeev, Margarita D. Samsonova, Yaroslav V. Dvoryanchikov, Tatiana V. Nikonova, Ekaterina V. Bondarenko, Marina Yu. Loguinova, Yulia A. Medvedeva, Dmitry N. Laptev, Rita I. Khusainova, Ildar R. Minniakhmetov, Marina V. Shestakova, Natalia G. Mokrysheva, Ivan I. Dedov
Abstract
Neurocognitive impairment is a prevalent co-morbidity in virologically suppressed people living with HIV (PLWH), yet the underlying mechanisms remain elusive and treatments lacking. We explored use of participant-derived directly induced neurons (iNs) to model neuronal biology and injury in PLWH. iNs retain age- and disease-related donor features, providing unique opportunities to reveal important aspects of neurological disorders. We obtained primary dermal fibroblasts from six virologically suppressed PLWH (range: 27-64 years, median: 53; 83% Male) and seven matched people without HIV (PWOH) (range: 27-66, median: 55; 71% Male). iNs were generated using transcription factors NGN2 and ASCL1, and validated by immunocytochemistry, single-cell-RNAseq, and electrophysiological recordings. Transcriptomic aging analyses confirmed retention of donor age-related signatures. Bulk-RNAseq identified 29 significantly differentially expressed genes between PLWH and PWOH iNs. Of these, 16 were downregulated and 13 upregulated in PLWH iNs. Protein-protein interaction network mapping indicates iNs from PLWH exhibit differences in extracellular matrix organization and synaptic transmission. IFI27 was upregulated in PLWH iNs, complementing independent post-mortem studies demonstrating elevated IFI27 expression in PLWH-derived brain tissue. FOXL2NB-FOXL2-LINC01391 expression was reduced in PLWH iNs and negatively correlated with neurocognitive impairment. Thus, we identified an iN gene signature of HIV revealing mechanisms of neurocognitive impairment in PLWH.
Authors
Philipp N. Ostermann, Youjun Wu, Scott Bowler, Samuel Martínez-Meza, Mohammad A. Siddiqui, David H. Meyer, Alberto Herrera, Brandon A. Sealy, Mega Sidharta, Kiran Ramnarine, Leslie Ann St. Bernard, Desiree Byrd, R. Jones, Masahiro Yamashita, Douglas F. Nixon, Lishomwa C. Ndhlovu, Ting Zhou, Teresa H. Evering
Abstract
Duchenne muscular dystrophy (DMD) is a fatal genetic muscle-wasting disease characterized by loss of dystrophin protein. Therapeutic attempts to restore a functional copy of dystrophin to striated muscle are under active development, and many utilize adeno-associated viral (AAV) vectors. However, the limited cargo capacity of AAVs precludes delivery of full-length dystrophin, a 427 kDa protein, to target tissues. Recently, we developed a novel method to express large dystrophin constructs using the protein trans-splicing (PTS) mechanism mediated by split inteins and myotropic AAV vectors. The efficacy of this approach to restore muscle function in mdx4cv mice was previously assessed using histology, dystrophin immunolabeling, and western blotting. Here, we expand our molecular characterization of dystrophin constructs with variable lengths using a mass spectrometry-based proteomics approach, providing insight into unique protein expression profiles in skeletal muscles of wild-type, dystrophic mdx4cv, and AAV-treated mdx4cv. Our data reveal several affected cellular processes in mdx4cv skeletal muscles with changes in the expression profiles of key proteins to muscle homeostasis, whereas successful expression of dystrophin constructs results in an intermediate to complete restoration. This study highlights several biomarkers that could be used in future preclinical or clinical studies to evaluate the effectiveness of therapeutic strategies.
Authors
Erynn E. Johnson, Theodore R. Reyes, Jeffrey S. Chamberlain, James M. Ervasti, Hichem Tasfaout
Abstract
The two main subgroups of autoimmune myasthenia gravis, a neuromuscular junction disorder associated with muscle weakness, are the early and late-onset forms, defined by onset before or after 50 years of age. Both carry acetylcholine-receptor autoantibodies, but differ in sex ratios, genetics and occurrence of disease-specific thymus inflammation. By applying multimodal techniques, including deep spectral cytometric phenotyping and single cell sequencing to peripheral blood and thymic lymphocyte samples we explored the possibility to discriminate the two forms by cellular immune phenotyping. Analyzing two independent cohorts we identified distinct immunological differences driven by three main lymphocyte populations. Lower frequencies of mucosa-associated invariant T cells and naïve CD8 T cells were observed in late-onset myasthenia, suggesting enhanced immune senescence. Further, a highly differentiated, canonical natural killer cell population was reduced in early-onset myasthenia, which was negatively correlated with the degree of thymic inflammation. Using only the frequency of these three populations, correct myasthenia subgroup assignment could be predicted with an accuracy of 90%. The NK cell population negatively associated to early-onset disease had a similar association to thymic hyperlasia, whereas the two T-cell populations point to enhanced immune senescence in late-onset myasthenia gravis. These distinct immunocellular endophenotypes for early- and late onset disease suggest differences in the immunopathogenic processes. Together with demographic factors and other disease subgroup-specific features, the frequency of the identified cell subpopulations may improve clinical classification, in turn of relevance for channeling to interventions.
Authors
Jakob Theorell, Nicolas Ruffin, Andrew Fower, Chiara Sorini, Philip Ambrose, Valentina Damato, Lahiru Handunnetthi, Isabel Leite, Sarosh R. Irani, Susanna Brauner, Adam E. Handel, Fredrik Piehl
Abstract
Over 95% of head and neck cancers are squamous cell carcinoma (HNSCC). HNSCC is mostly diagnosed late, causing a poor prognosis despite the application of invasive treatment protocols. Tumor-educated platelets (TEPs) have been shown to hold promise as a molecular tool for early cancer diagnosis. We sequenced platelet mRNA isolated from blood of 101 HNSCC patients and 101 propensity-score matched non-cancer controls. Two independent machine learning classification strategies were employed using a training and validation approach to identify a cancer predictor: a particle swarm optimized support vector machine (PSO-SVM) and a least absolute shrinkage and selection operator (LASSO) logistic regression model. The best performing PSO-SVM predictor consisted of 245 platelet transcripts and reached a maximum area under the curve (AUC) of 0.87. For the LASSO-based prediction model 1,198 mRNAs were selected, resulting in an median AUC of 0.84, independent of HPV status. Our data show that TEP RNA classification by different AI tools is promising in the diagnosis of HNSCC.
Authors
N.E. Wondergem, J.B. Poell, S.G.J.G In 't Veld, E. Post, S.W. Mes, M.G. Best, W.N. van Wieringen, T. Klausch, R.J. Baatenburg de Jong, C.H.J. Terhaard, R.P. Takes, J.A. Langendijk, I.M. Verdonck-de Leeuw, F. Lamers, C.R. Leemans, E. Bloemena, T. Würdinger, R.H Brakenhoff


