
Abstract
Actinomycetes have produced a variety of bioactive secondary metabolites; however, discovering new actinobacterial natural products using conventional approaches has become increasingly challenging. Meanwhile, genomic studies of actinomycetes have revealed that numerous secondary metabolite biosynthetic gene clusters (SM-BGCs) remain untapped. Thus, utilizing these secondary metabolic pathways is expected to facilitate the discovery of new actinomycetes-derived natural products. In this review, I primarily describe our research on the utilization of these untapped actinobacterial SM-BGCs and the discovery of new secondary metabolites. First, I introduce our studies on the activation of silent SM-BGCs through the co-cultivation of various actinomycetes with mycolic acid-containing bacteria (MACB), which led to the identification of 20 actinobacterial secondary metabolites, including 16 new compounds. In the latter part, I describe our recent findings on arsenic-related secondary metabolism, which has been overlooked in model actinomycetes, including the identification of a novel organoarsenic natural product, and the elucidation of its unique biosynthetic strategy, which is independent of S-adenosylmethionine (SAM)-dependent enzymes.
Graphical abstract

Similar content being viewed by others
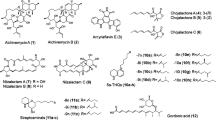
Introduction
Bacterial secondary metabolites often exhibit remarkable bioactivities and have significantly contributed to human society through their application in pharmaceuticals, agrochemicals and food additives. In particular, since the discovery of actinomycins and streptomycin [1, 2], numerous bioactive secondary metabolites have been identified from actinomycetes [3–5]. However, the continuous exploration of actinomycetes has made the discovery of new natural products increasingly challenging.
Meanwhile, genomic studies have revealed that actinomycetes harbor numerous secondary metabolite biosynthetic gene clusters (SM-BGCs) [6–8], far exceeding the total number of actinobacterial secondary metabolites identified to date. Thus, the majority of SM-BGCs remain cryptic under standard culture conditions, and activating these untapped actinobacterial SM-BGCs is expected to facilitate the discovery of new bioactive compounds. In fact, new secondary metabolites have been successfully identified through the activation of cryptic SM-BGCs using heterologous expression [9, 10], the application of chemical elicitors [11, 12], and genetic manipulation [13].
Furthermore, secondary metabolism involving elements that are rarely found in natural products is also noteworthy. For example, some actinomycetes possess specific SM-BGCs and produce organofluorine [14–16] and organoselenium metabolites [17], representing an extremely rare class of natural products. However, a large portion of these SM-BGCs is likely overlooked, even if they are functional, because conventional culture media typically lack the necessary elements for their biosynthesis.
Under these circumstances, we have continued our efforts to discover new natural products by utilizing untapped actinobacterial SM-BGCs. In this review, we first describe our studies on the activation of actinobacterial SM-BGCs through co-culture with mycolic acid-containing bacteria (MACB). In the latter part, we present our recent findings on actinobacterial secondary metabolism related to organoarsenical natural products.
Combined-culture: effective activation of actinobacterial SM-BGCs through co-culture of actinomycetes with MACB
Co-culture is also an effective approach for activating cryptic SM-BGCs, and several co-culture studies have successfully led to the isolation of new microbial natural products [18–20]. It is a simple and broadly applicable method that does not require genetic manipulation or expensive reagents. However, conventional co-culture approaches often require extensive screening to identify suitable microbial combinations. To address this limitation, Onaka et al. proposed an efficient co-culture strategy using MACB as a partner to activate cryptic SM-BGCs in actinomycetes, which they termed "combined-culture" [21].
Initially, they conducted a screening of bacteria capable of activating actinobacterial SM-BGCs using Streptomyces lividans TK23 as an indicator, which produces pigmented antibiotics (actinorhodin and undecylprodigiosin) in response to specific stimulation. As a result, Tsukamurella pulmonis TP-B0596 (T. pulmonis) was identified as an effective inducer of pigmented antibiotics production in S. lividans TK23, both on agar plates (Fig. 1a) and in liquid cultures (Fig. 1b) [21].

a Production of pigmented antibiotics in Streptomyces lividans TK23 (S. lividans) was induced by co-culturing with Tsukamurella pulmonis TP-B0596 (T. pulmonis) on an agar plate. b Production of pigmented antibiotics in S. lividans was also induced in liquid culture through co-cultivation with T. pulmonis. c Chemical structures of alchivemycins (1 and 2), with the 2H-tetrahydro-4,6-dioxo-1,2-oxazine moiety highlighted in red
The genus Tsukamurella belongs to MACB, a group of bacteria characterized by the presence of mycolic acid on their cell surface. Notably, other MACB genera, such as Corynebacterium, Dietzia, and Rhodococcus, also induced pigment production in S. lividans TK23. The importance of mycolic acid was further supported by the finding that Corynebacterium glutamicum Δpks13, a mutant lacking the mycolic acid biosynthetic gene, lost its inducing activity [21].
More importantly, MACB not only induced pigment production in S. lividans TK23 but also broadly activated SM-BGCs in various actinomycetes. Onaka et al. reported that co-culturing T. pulmonis with 112 strains of soil-derived Streptomyces resulted in metabolic profile changes in 97 strains, among which 35 strains exhibited enhanced secondary metabolite production [21]. In particular, two novel polyketides featuring a unique 2H-tetrahydro-4,6-dioxo-1,2-oxazine moiety, named alchivemycins A (1) and B (2), were isolated from Streptomyces sp. TP-A0867 when co-cultured with T. pulmonis (Fig. 1c) [21–23]. Alchivemycins exhibited potent and selective antibacterial activity against Kocuria rhizophila ATCC 9341 (formerly Micrococcus luteus ATCC 9341), with MIC values of 0.03 µg/ml for 1 and 0.004 µg/ml for 2 [23].
Application of combined-culture strategy to Streptomyces strains
Encouraged by the discovery of alchivemycins, which possess unique chemical structures and potent bioactivity, we aimed to obtain new secondary metabolites through the combined-culture strategy. Initially, we targeted Streptomyces strains, a representative genus of actinomycetes that has produced the majority of actinobacterial secondary metabolites [3, 24]. In our combined-culture screening, T. pulmonis was consistently used as the MACB, and changes in the metabolic profile were monitored by HPLC equipped with a diode array detector (HPLC–DAD). Thus far, we have isolated eleven secondary metabolites, including seven new compounds, from four Streptomyces strains as described below (3−13, Fig. 2a).

a Chemical structures of secondary metabolites (3–13) obtained from Streptomyces strains through our combined-culture screening. b Chemical synthesis of possible stereoisomers of 6 and 7. The figure illustrates the case in which (R)-14 was used as the starting material. c Chemical structure of tripartilactam (17), as originally proposed by Oh et al. in 2012 [33]
Streptomyces cinnamoneus NBRC 13823 was found to produce three pigmented metabolites, whose production was significantly enhanced in the presence of MACB [25]. Based on chromatographic purification and spectroscopic analysis, one of these metabolites was identified as a new indolocarbazole alkaloid, named arcyriaflavin E (3), while the remaining two were the known indolocarbazole alkaloids arcyriaflavin A (4) and BE-13793C (5) [26, 27]. 3 and 5 exhibited moderate cytotoxicity against P388 murine leukemia cells, with IC50 values of 39 and 33 µM, respectively, while 4 showed no cytotoxicity up to 100 µM [25].
Streptomyces sp. CJ-5, which we isolated from a soil sample collected in Isumi City (Chiba, Japan), produced three unknown metabolites exclusively in the presence of MACB. Our detailed spectroscopic analysis revealed that all of these metabolites are new butanolides, named chojalactones A–C (6−8) [28]. The relative configurations of 6 and 7 were determined by NMR analysis, while their absolute configurations were established through the chemical synthesis of all possible enantiomers from 3-methyl-γ-butyrolactone (14) (Fig. 2b).
Initially, 14 was condensed with triene acid chloride (15) via Ti-crossed Claisen condensation using N-methylimidazole as a catalyst [29], yielding the β-ketolactone (16). 16 was then subjected to Pd/C-catalyzed direct α-oxidization under an O2 atmosphere [30] to obtain the desired stereoisomers: (2R, 3S) and (2S, 3S) enantiomers from (3R)-16, and (2S, 3R) and (2R, 3R) enantiomers from (3S)-16. By comparing the retention times of the natural products with those of synthetic standards using chiral-phase HPLC, the absolute configurations of 6 and 7 were determined to be (2R, 3S) and (2R, 3R), respectively. The absolute configuration at the C-2 position of 8 was deduced to be R based on its consistency with 6 and 7. Finally, 6 and 7 exhibited moderate cytotoxicity against P388 murine leukemia cells, with IC50 values of 28 and 18 µM, respectively.
It was found that Streptomyces sp. NZ-6, which we isolated from a soil sample collected in Niiza City (Saitama, Japan), produces three metabolites in a MACB-dependent manner. Two of them were identified as new polyene macrolactams with a common [5, 23]-bicyclic skeleton and were named niizalactams A (9) and B (10) [31]. The relative and absolute configurations of 9 and 10 were fully determined based on exhaustive NMR analysis of the natural products and their synthetic derivatives, including the application of the modified Mosher’s method to secondary alcohols [32].
The NMR spectrum of the remaining metabolite closely matched that of tripartilactam (17), a polyene macrolactam with an [4, 8, 18]-tricyclic skeleton, which was isolated from Streptomyces sp. SNA112 by Oh et al. in 2012 (Fig. 2c) [33]. However, based on our thorough NMR analysis, the presence of an [6, 6, 18]-tricyclic skeleton was supported, leading to its designation as niizalactam C (11). Recently, Oh et al. conducted a re-investigation of the chemical structure of 17, including a 13C–13C COSY analysis of a 13C-labeled derivative of 17 [34]. In their study, the original structure of 17, proposed in 2012, was revised to be identical to 11, as proposed by our group.
Along with the above examples, we also found that Streptomyces sp. TAKO-2, which we isolated from a soil sample collected in Tako Town (Chiba, Japan), produced compounds with absorption in the long-wavelength region (> 400 nm) when co-cultured with MACB [35]. The spectral data of two of these metabolites matched those of the known aromatic polyketides, julichrome Q8,8 (12) [36] and julichrome Q6 (13) [37]. Considering the similarity of their UV–vis spectra, the remaining metabolites were also inferred to be related analogs to 12 and 13.
Application of combined-culture screening to non-Streptomyces actinomycetes
As describe above, our combined-culture screening initially targeted Streptomyces strains; however, we later shifted our focus to other actinomycete genera. Non-Streptomyces actinomycetes, often referred to as rare actinomycetes, have been relatively unexplored for secondary metabolites compared to Streptomyces. However, recent genomic studies of non-Streptomyces genera have revealed the presence of SM-BGCs comparable to or even exceeding those in Streptomyces [7, 8]. In fact, a considerable number of new bioactive secondary metabolites have been identified from these genera [5, 38]. Thus far, through combined-culture screening, we have identified nine new secondary metabolites from four non-Streptomyces actinomycetes (18–26, Fig. 3a).

a Chemical structures of secondary metabolites (18–26) obtained from non-Streptomyces strains through our combined-culture screening. b 18 spontaneously converts to 19 in DMSO-d6 solution, accompanied by double bond isomerization. c. Non-enzymatic isomerization of 21, leading to the formation of the 3-methyl-isoxazolidinone scaffold in 20
The genus Umezawaea is a relatively new actinomycete genus proposed in 2007 [39], and no reports on its secondary metabolites had been published until we started the combined-culture screening. Umezawaea sp. RD066910 does not produce any notable metabolites under pure-culture conditions; however, we found that it produced two metabolites with similar UV spectra when co-cultured with MACB. Spectral analysis of the purified metabolites revealed that both are new polycyclic tetramate macrolactams and represent the first secondary metabolites identified from the genus Umezawaea. We named them umezawamides A (18) and B (19), which are structural isomers differing only in the position of the carbon–carbon double bonds [40].
During NMR analysis, it was found that 18 gradually converted to 19 in DMSO-d6 at room temperature, suggesting that 19 is an artifact generated from 18 (Fig. 3b). The absolute configurations of 18 and 19, except at the C-16 position, were fully elucidated by combining NMR analysis, conformational search using the Merck Molecular Force Field (MMFF), and ECD calculations. 18 and 19 exhibited cytotoxicity against P388 murine leukemia cells, with IC50 values of 3.7 and 4.8 µM, respectively. Additionally, 18 showed antifungal activity against Candida albicans, whereas 19 did not [40].
Catenuloplanes sp. RD067331 exhibited a significant increase in the production of two metabolites when cultured in the presence of MACB. Spectral analysis revealed that these metabolites are new heterocyclic peptides, which we named catenulobactins A (20) and B (21) [41]. Total acid hydrolysis, followed by chiral-phase GC–MS analysis of 21, revealed that N5-hydroxyornithine is in the d-form, and that both 5-methyl-oxazolines (MeOzn) are derived from l-threonine, leading to the complete determination of its absolute configuration.
20 is a structural isomer of 21, featuring a unique 3-methyl-isoxazolidinone ring in place of one of the MeOzn moieties. Considering the biosynthetic mechanism of acinetobactin proposed by Wencewicz et al. [42], the 3-methyl-isoxazolidinone ring in 20 is presumed to be generated through an intramolecular rearrangement of 21, involving the MeOzn moiety and the adjacent N–OH group (Fig. 3c). Notably, we found that 21 spontaneously converted to 20 when incubated in phosphate buffer (pH 7.0) at room temperature [41]. Finally, we found that 21 exhibits Fe(III)-chelating activity as well as cytotoxicity against P388 murine leukemia cells, with an IC50 value of 22.4 µM, whereas 20 exhibited neither activity [41].
Micromonospora wenchangensis HEK-797, isolated from lake sediment at Hegura Island (Ishikawa, Japan), produced two metabolites specifically in the presence of MACB. NMR analysis of the purified metabolites revealed that one possesses a unique [5, 5, 6, 16]-tetracyclic skeleton, which we named dracolactam A (22), while the other is a new bicyclic lactam featuring a THF ring, named dracolactam B (23) [43]. The relative and absolute configurations of 22 and 23 were determined through NOESY analysis and a careful evaluation of vicinal coupling constants of the natural products, combined with detailed NMR analysis of the corresponding acetonide derivatives and the application of the modified Mosher’s method.
Epoxidation-mediated intramolecular cyclization of polyene macrolactams triggered by combined-culture
Considering the positions of functional groups, 22 and 23 were presumed to originate from a common 26-membered polyene macrolactam (27), whose planar structure is identical to those of micromonolactam [44] and isomicromonolactam [45] (Fig. 4a). In our proposed mechanism, the double bond between C-22 and C-23 of 27 is initially epoxidized, generating an unstable intermediate, which subsequently undergoes intramolecular cyclization.

a Proposed biosynthetic pathways of dracolactams (22 and 23) and mirilactams C–E (24–26), involving epoxidation-mediated intramolecular cyclization of 26-membered macrolactam precursors. b Proposed biosynthetic pathway of niizalactam A (9) from a 26-membered sceliphrolactam-type precursor (28). Niizalactam B (10) is also presumed to be biosynthesized from the C-10 deoxy derivative of 28. c Chemical structures of ciromicins A (29) and B (30). d Chemical structures of heronamides (32–35)
If the amide nitrogen directly attacks the epoxide ring at C-22, the resulting bicyclic compound undergoes further intramolecular Diels–Alder reaction, forming 22 (Fig. 4a, Path A). Meanwhile, if the hydroxyl group at C-11 attacks the C-14 carbon, inducing a 1,10-addition accompanied by epoxide ring opening, 23 is formed (Fig. 4a, Path B). Notably, M. wenchangensis HEK-797 produced a metabolite with a UV–Vis spectrum and molecular formula identical to 27 under both pure-culture and combined-culture conditions, suggesting that MACB specifically activates the epoxidation of 27 rather than its biosynthesis [43].
A similar biosynthetic strategy can also be applied to 9 and 10, in which a 26-membered polyene macrolactam (28), sharing the same planar structure as sceliphrolactam [46], or its C-10 deoxy derivative, is presumed to be the precursor (Fig. 4b). It was assumed that epoxidation at the C-22 and C-23 double bond, followed by nucleophilic pyrrolidinol formation, leads to the generation of 9 and 10 (Fig. 4b). Additionally, Bachmann et al. reported that the rare actinomycete Nocardiopsis sp. FU40 ΔApoS produced ciromicins A (29) and B (30) (Fig. 4c) strictly in the presence of MACB (Rhodococcus wratislaviensis) [47]. 29 and 30, which contain the pyrrolidinol moiety, are proposed to be biosynthesized through the epoxidation of a 22-membered glycosylated polyene macrolactam, followed by intramolecular cyclization [47].
Based on these findings, we hypothesized that applying combined-culture could activate cryptic epoxidation-mediated cyclization pathways in polyene macrolactam-producing actinomycetes. To validate this hypothesis, we focused on Actinosynnema mirum NBRC 14064 (A. mirum), which is known to produce a 26-membered polyene macrolactam named mirilactam A (31) (Fig. 4a) [48].
We confirmed that A. mirum produces 31 under monoculture conditions, and as expected, specifically produced three additional metabolites when co-cultured with MACB [49]. Detailed structural analysis of the purified metabolites revealed that they are all new compounds derived from 31, which we named mirilactams C–E (24−26, Fig. 3a) [49]. The biosynthetic mechanism of 24 and 25 is similar to that of 22 and 23. Meanwhile, in the biosynthesis of 26, it is likely that the hydroxyl group at C-10 in the epoxidized intermediate attacks the C-14 carbon, leading to the formation of a tetrahydropyran ring (Fig. 4a, Path C).
Polycyclic metabolites, likely generated by the epoxidation of polyene macrolactams, have also been found in the mono-culture of actinomycetes. Heronamide A (32) is a [5, 5, 6, 10]-tetracyclic metabolite containing a pyrrolidinol moiety, identified from the marine-derived Streptomyces sp. CMB-M0406 (Fig. 4d) [50, 51], while heronamide D (33), which shares the same tetracyclic skeleton as 32, was identified from the deep-sea-derived Streptomyces sp. SCSIO 03032 (Fig. 4d) [52].
Strains CMB-M0406 and SCSIO 03032 also produce 20-membered polyene macrolactams, named heronamides C (34) and F (35), respectively [50, 52, 53] (Fig. 4d), which are likely precursors of 32 and 33. Notably, the production of 32 significantly increased when artificial seawater was added to the culture medium, whereas no difference was observed in the production of 34 between saline and non-saline conditions [50]. Additionally, the production of 33 was observed in the medium containing artificial seawater [52], suggesting that salt stress induce the epoxidation of polyene macrolactams in certain actinomycetes.
To the best of my knowledge, no epoxidase genes have been clearly identified within the polyene macrolactam BGCs. Thus, it is possible that an epoxidase gene located outside the polyene macrolactam BGC is activated during co-cultivation with MACB. On the other hand, it has been reported that 34 undergoes a non-enzymatic conversion to 32 in DMSO at room temperature [53], raising the possibility that the MACB-induced epoxidation could occur through a non-enzymatic mechanism, such as the generation of reactive oxygen species.
Identification and biosynthetic studies of bisenarsan: a novel organoarsenic secondary metabolite produced by model actinomycetes
Organoarsenic compounds containing C–As bonds are quite rare among bacterial natural products. A representative example is a series of simple metabolites biosynthesized through the sequential methylation of arsenite [As(III)], often followed by oxidation, a process widely recognized as a detoxification mechanism for highly toxic inorganic arsenic [54–56]. In other examples, several cyanobacteria have been reported to produce oxo-arsenosugars [57, 58], while a potent glutamine synthetase inhibitor, arsinothricin, has been identified from the terrestrial bacterium Burkholderia gladioli GSRB05 [59, 60].
In the biosynthesis of these organoarsenic natural products in bacteria, all C–As bonds are formed by S-adenosylmethionine (SAM)-dependent enzymes, among which SAM-dependent arsenic methyltransferases are widely distributed in bacterial genomes [54–56]. In addition, recent studies have shown that radical SAM enzymes are also involved in C–As bond formation. The adenosylation of arsenic in oxo-arsenosugar biosynthesis [61] and the 3-amino-3-carboxypropyl radical transfer to the arsenic atom in arsinothricin biosynthesis [62] are both catalyzed by radical SAM enzymes.
Despite their high capacity for secondary metabolite production, actinomycetes have long been known to produce only simple methylated arsenicals [56]. However, in 2016, Cruz-Morales et al. reported that the two model actinomycetes Streptomyces lividans 1326 and Streptomyces coelicolor A3(2) produce an unknown organoarsenic metabolite (36, C14H27AsO5) [63]. They further proposed that a conserved SM-BGC among these strains, which we later termed the bsn cluster, is responsible for the biosynthesis of 36 (Fig. 5a) [63].

a Schematic representation of the SM-BGC for 36 in Streptomyces lividans 1326 (bsn cluster). b Chemical structure of 36, along with its proposed components (37 and 38). c Proposed biosynthetic pathway of 36 from As(V)
The bsn cluster includes genes involved in arsenic resistance and arsenic-dependent transcriptional regulation. In fact, it has been reported that the transcription of several genes within the bsn cluster is significantly upregulated under exposure to arsenate [As(V)] [63]. More importantly, the bsn cluster does not contain any genes encoding SAM-dependent enzymes. However, it has been reported that disruption of the 5-enolpyruvylshikimate-3-phosphate (EPSP) synthase homolog (bsnM) completely abolished the production of 36 [63].
Based on these findings, 36 was expected to be an organoarsenic natural product biosynthesized through C–As bond formation without the involvement of SAM-dependent enzymes. Therefore, we aimed to determine the chemical structure of 36. Initially, S. lividans 1326 was mass-cultured in a medium containing As(V) as the sole arsenic source, but only a trace amount of 36 (< 0.1 mg) was obtained [64]. However, 1H NMR and HR-MS/MS analysis of the trace purified material suggested that 36 is an O-acyl derivative of (2-hydroxyethyl)arsonic acid (2-HEA, 37) (Fig. 5b).
In addition, Wang et al. reported that heterologous expression of the polyketide synthase (PKS) genes (bsnPKS and bsnFB, Fig. 5a) resulted in the production of 2,4,6-trimethyl-2-nonenoic acid (2,4,6-TMNA, 38) [65], suggesting that the acyl group of 36 is derived from 38 (Fig. 5b). Notably, when chemically synthesized 37 was added to the culture medium, the production of 36 in S. lividans 1326 significantly increased. Finally, NMR re-analysis of the purified metabolite confirmed that the chemical structure of 36 matched our initial proposal, and we named it bisenarsan (Fig. 5b) [64].
Through gene disruption in S. lividans 1326, we revealed that, in addition to bsnM (EPSP synthase) [63], genes encoding phosphoglycerate mutase (bsnN) and phosphonopyruvate decarboxylase (bsnJ) are also required for the biosynthesis of 36 (Fig. 5a) [64]. Furthermore, supplementation with chemically synthesized 37 in the culture medium of all the disruptants of bsnM, bsnN, and bsnJ restored the production of 36, suggesting that these genes are responsible for the biosynthesis of 37 [64].
Based on these observation as well as the predicted functions of bsn genes, we propose the biosynthetic pathway of 36 as follows (Fig. 5c). Initially, the pyruvate group in EPSP, presumably derived from the shikimate pathway, is transferred to As(V) by BsnM, resulting in the formation of arsenoenolpyruvate (AEP, 39). 39 is then converted to arsonopyruvate (AnPy, 40) by BsnN, accompanied by C–As bond formation, and subsequently decarboxylated to arsonoacetoaldehyde (AnAA, 41) by BsnJ. 41 is presumed to be reduced to 37 by reductases encoded within the bsn cluster or by an orphan reductase in S. lividans 1326. Finally, 38, bound to the acyl carrier protein of the PKS module (BsnPKS and BsnFB), is thought to undergo nucleophilic attack by 37, leading to the release of 36. It has been reported that the expression of bsnPKS is upregulated upon exposure to As(V) [63], suggesting that ester bond formation between 37 and 38 is part of the genuine biosynthetic pathway rather than a shunt pathway.
The biological function of 36 remains unclear; however, we have previously shown that its toxicity is significantly lower than that of As(V) [64], suggesting that the biosynthesis of 36 may serve as a detoxification mechanism for inorganic arsenic. Additionally, we have demonstrated that 37 exhibits stronger antibacterial activity against Staphylococcus aureus than As(V) [64], raising the possibility that 36 is produced as “Trojan horse” antibiotic, with 37 serving as the active form.
Intriguingly, the gene set bsnMN, responsible for the conversion of inorganic arsenic [As(V)] to 40, is widely distributed among actinobacterial genomes, whereas the surrounding regions exhibit considerable genetic diversity [63, 64].
Conclusion
As described earlier, we identified a total of 20 actinobacterial secondary metabolites, including 16 new compounds, from Streptomyces strains (3−13, Fig. 2a) and non-Streptomyces genera (18−26, Fig. 3a) by activating cryptic SM-BGCs through co-culture with MACB (combined-culture). The continued application of the combined-culture strategy to other actinomycetes is expected to facilitate the discovery of additional new bioactive natural products. In fact, the number of new secondary metabolites obtained through this strategy has continued to increase in recent years [66–69].
Furthermore, we isolated and determined the chemical structure of a novel organoarsenic metabolite, named bisenarsan (36), produced by two model actinomycetes (Fig. 5b). Additionally, our study provided significant insight into the biosynthesis of 36, suggesting that C–As bond in 36 is likely generated through a novel pathway that does not involve SAM-dependent enzymes (Fig. 5c). Notably, the bsnMN homologs, presumably responsible for the generation of AnPy (40) from As(V), are widely distributed among actinobacterial genomes, and genome mining targeting these homologs is expected to facilitate the discovery of new organoarsenic metabolites that share 40 as a common precursor.
Overall, a large number of untapped secondary metabolic pathways still exist in actinomycetes, and we continue to explore and utilize these pathways to discover new bioactive secondary metabolites.
Change history
11 June 2025
A Correction to this paper has been published: https://doi.org/10.1007/s11418-025-01922-6
References
Waksman SA, Woodruff HB (1940) Bacteriostatic and bactericidal substances produced by a soil actinomyces. Exp Biol Med 45:609–614. https://doi.org/10.3181/00379727-45-11768
Schatz A, Bugle E, Waksman SA (1944) Streptomycin, a substance exhibiting antibiotic activity against Gram-positive and Gram-negative bacteria. Exp Biol Med 55:66–69. https://doi.org/10.3181/00379727-55-14461
Bérdy J (2005) Bioactive microbial metabolites. J Antibiot 58:1–26. https://doi.org/10.1038/ja.2005.1
Jakubiec-Krzesniak K, Rajnisz-Mateusiak A, Guspiel A, Ziemska J, Solecka J (2018) Secondary metabolites of actinomycetes and their antibacterial, antifungal and antiviral properties. Pol J Microbiol 67:259–272. https://doi.org/10.21307/pjm-2018-048
Igarashi Y (2023) Development of a drug discovery approach from microbes with a special focus on isolation sources and taxonomy. J Antibiot 76:365–383. https://doi.org/10.1038/s41429-023-00625-y
Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M (2001) Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci USA 98:12215–12220. https://doi.org/10.1073/pnas.211433198
Doroghazi JR, Albright JC, Goering AW, Ju KS, Haines RR, Tchalukov KA, Labeda DP, Kelleher NL, Metcalf WW (2014) A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol 10:963–968. https://doi.org/10.1038/nchembio.1659
Jorgensen TS, Mohite OS, Sterndorff EB, Alvarez-Arevalo M, Blin K, Booth TJ, Charusanti P, Faurdal D, Hansen TO, Nuhamunada M, Mourched AS, Palsson BO, Weber T (2024) A treasure trove of 1034 actinomycete genomes. Nucleic Acids Res 52:7487–7503. https://doi.org/10.1093/nar/gkae523
Kozakai R, Ono T, Hoshino S, Takahashi H, Katsuyama Y, Sugai Y, Ozaki T, Teramoto K, Teramoto K, Tanaka K, Abe I, Asamizu S, Onaka H (2020) Acyltransferase that catalyses the condensation of polyketide and peptide moieties of goadvionin hybrid lipopeptides. Nat Chem 12:869–877. https://doi.org/10.1038/s41557-020-0508-2
Nishimura T, Kudo K, Izumikawa M, Kozone I, Hashimoto J, Kagaya N, Suenaga H, Takeuchi K, Shin-Ya K (2024) Isolation and structure elucidation of JBIR-157, a skeletally novel aromatic polyketide produced by the heterologous expression of a cryptic gene cluster. Chem Pharm Bull 72:475–479. https://doi.org/10.1248/cpb.c24-00144
Xu F, Wu Y, Zhang C, Davis KM, Moon K, Bushin LB, Seyedsayamdost MR (2019) A genetics-free method for high-throughput discovery of cryptic microbial metabolites. Nat Chem Biol 15:161–168. https://doi.org/10.1038/s41589-018-0193-2
Han EJ, Lee SR, Hoshino S, Seyedsayamdost MR (2022) Targeted discovery of cryptic metabolites with antiproliferative activity. ACS Chem Biol 17:3121–3130. https://doi.org/10.1021/acschembio.2c00588
Du D, Katsuyama Y, Onaka H, Fujie M, Satoh N, Shin-Ya K, Ohnishi Y (2016) Production of a novel amide-containing polyene by activating a cryptic biosynthetic gene cluster in Streptomyces sp. MSC090213JE08. ChemBioChem 17:1464–1471. https://doi.org/10.1002/cbic.201600167
Sanada M, Miyano T, Iwadare S, Williamson JM, Arison BH, Smith JL, Douglas AW, Liesch JM, Inamine E (1986) Biosynthesis of fluorothreonine and fluoroacetic acid by the thienamycin producer, Streptomyces cattleya. J Antibiot 39:259–265. https://doi.org/10.7164/antibiotics.39.259
Deng H, Ma L, Bandaranayaka N, Qin Z, Mann G, Kyeremeh K, Yu Y, Shepherd T, Naismith JH, O’Hagan D (2014) Identification of fluorinases from Streptomyces sp MA37, Norcardia brasiliensis, and Actinoplanes sp N902-109 by genome mining. ChemBioChem 15:364–368. https://doi.org/10.1002/cbic.201300732
Zhu XM, Hackl S, Thaker MN, Kalan L, Weber C, Urgast DS, Krupp EM, Brewer A, Vanner S, Szawiola A, Yim G, Feldmann J, Bechthold A, Wright GD, Zechel DL (2015) Biosynthesis of the fluorinated natural product nucleocidin in Streptomyces calvus is dependent on the bldA-specified Leu-tRNAUUA molecule. ChemBioChem 16:2498–2506. https://doi.org/10.1002/cbic.201500402
Kayrouz CM, Huang J, Hauser N, Seyedsayamdost MR (2022) Biosynthesis of selenium-containing small molecules in diverse microorganisms. Nature 610:199–204. https://doi.org/10.1038/s41586-022-05174-2
Cueto M, Jensen PR, Kauffman C, Fenical W, Lobkovsky E, Clardy J (2001) Pestalone, a new antibiotic produced by a marine fungus in response to bacterial challenge. J Nat Prod 64:1444–1446. https://doi.org/10.1021/np0102713
Oh DC, Kauffman CA, Jensen PR, Fenical W (2007) Induced production of emericellamides A and B from the marine-derived fungus Emericella sp. in competing co-culture. J Nat Prod 70:515–520. https://doi.org/10.1021/np060381f
Moussa M, Ebrahim W, Bonus M, Gohlke H, Mandi A, Kurtan T, Hartmann R, Kalscheuer R, Lin W, Liu Z, Proksch P (2019) Co-culture of the fungus Fusarium tricinctum with Streptomyces lividans induces production of cryptic naphthoquinone dimers. RSC Adv 9:1491–1500. https://doi.org/10.1039/c8ra09067j
Onaka H, Mori Y, Igarashi Y, Furumai T (2011) Mycolic acid-containing bacteria induce natural-product biosynthesis in Streptomyces species. Appl Environ Microbiol 77:400–406. https://doi.org/10.1128/AEM.01337-10
Igarashi Y, Kim Y, In Y, Ishida T, Kan Y, Fujita T, Iwashita T, Tabata H, Onaka H, Furumai T (2010) Alchivemycin A, a bioactive polycyclic polyketide with an unprecedented skeleton from Streptomyces sp. Org Lett 12:3402–3405. https://doi.org/10.1021/ol1012982
Kim Y, In Y, Ishida T, Onaka H, Igarashi Y (2013) Biosynthetic origin of alchivemycin A, a new polyketide from Streptomyces and absolute configuration of alchivemycin B. Org Lett 15:3514–3517. https://doi.org/10.1021/ol401071j
Watve MG, Tickoo R, Jog MM, Bhole BD (2001) How many antibiotics are produced by the genus Streptomyces? Arch Microbiol 176:386–390. https://doi.org/10.1007/s002030100345
Hoshino S, Zhang L, Awakawa T, Wakimoto T, Onaka H, Abe I (2015) Arcyriaflavin E, a new cytotoxic indolocarbazole alkaloid isolated by combined-culture of mycolic acid-containing bacteria and Streptomyces cinnamoneus NBRC 13823. J Antibiot 68:342–344. https://doi.org/10.1038/ja.2014.147
Horton PA, Longley RE, McConnell OJ, Ballas LM (1994) Staurosporine aglycone (K252-c) and arcyriaflavin A from the marine ascidian, Eudistoma sp. Experientia 50:843–845. https://doi.org/10.1007/BF01956468
Kojiri K, Kondo H, Yoshinari T, Arakawa H, Nakajima S, Satoh F, Kawamura K, Okura A, Suda H, Okanishi M (1991) A new antitumor substance BE-13793C, produced by a streptomycete. Taxonomy, fermentation, isolation, structure determination and biological activity. J Antibiot 44:723–728. https://doi.org/10.7164/antibiotics.44.723
Hoshino S, Wakimoto T, Onaka H, Abe I (2015) Chojalactones A-C, cytotoxic butanolides isolated from Streptomyces sp. cultivated with mycolic acid containing bacterium. Org Lett 17:1501–1504. https://doi.org/10.1021/acs.orglett.5b00385
Misaki T, Nagase R, Matsumoto K, Tanabe Y (2005) Ti-crossed-Claisen condensation between carboxylic esters and acid chlorides or acids: a highly selective and general method for the preparation of various beta-keto esters. J Am Chem Soc 127:2854–2855. https://doi.org/10.1021/ja043833o
Sajiki H, Monguchi Y, Takahashi T, Iida Y, Fujiwara Y, Inagaki Y, Maegawa T (2008) Pd/C-Catalyzed direct α-oxygenation of 1,3-dicarbonyl compounds using molecular oxygen. Synlett 2008:2291–2294. https://doi.org/10.1055/s-2008-1078023
Hoshino S, Okada M, Wakimoto T, Zhang H, Hayashi F, Onaka H, Abe I (2015) Niizalactams A-C, multicyclic macrolactams isolated from combined culture of Streptomyces with mycolic acid-containing bacterium. J Nat Prod 78:3011–3017. https://doi.org/10.1021/acs.jnatprod.5b00804
Ohtani I, Kusumi T, Kashman Y, Kakisawa H (1991) High-field FT NMR application of Mosher’s method. The absolute configuration of marine terpenoids. J Am Chem Soc 113:4092–4096. https://doi.org/10.1021/ja00011a006
Park SH, Moon K, Bang HS, Kim SH, Kim DG, Oh KB, Shin J, Oh DC (2012) Tripartilactam, a cyclobutane-bearing tricyclic lactam from a Streptomyces sp. in a dung beetle’s brood ball. Org Lett 14:1258–1261. https://doi.org/10.1021/ol300108z
Hwang S, Kim E, Lee J, Shin J, Yoon YJ, Oh DC (2020) Structure revision and the biosynthetic pathway of tripartilactam. J Nat Prod 83:578–583. https://doi.org/10.1021/acs.jnatprod.9b00819
Hoshino S, Okada M, Onaka H, Abe I (2016) Effective production of aromatic polyketides in Streptomyces using a combined-culture method. Nat Prod Commun 11:979–981. https://doi.org/10.1177/1934578x1601100727
Tsuji N, Nagashima K (1970) Studies on julimycins—VIII: the structures of julichromes Q1·7, Q8·8, Q3·8, Q3·3 and Q1·9. Tetrahedron 26:5201–5213. https://doi.org/10.1016/s0040-4020(01)98729-0
Prag A, Gruning BA, Hackh M, Ludeke S, Wilde M, Luzhetskyy A, Richter M, Luzhetska M, Gunther S, Muller M (2014) Regio- and stereoselective intermolecular oxidative phenol coupling in Streptomyces. J Am Chem Soc 136:6195–6198. https://doi.org/10.1021/ja501630w
Subramani R, Aalbersberg W (2013) Culturable rare Actinomycetes: diversity, isolation and marine natural product discovery. Appl Microbiol Biotechnol 97:9291–9321. https://doi.org/10.1007/s00253-013-5229-7
Labeda DP, Kroppenstedt RM (2007) Proposal of Umezawaea gen. nov., a new genus of the Actinosynnemataceae related to Saccharothrix, and transfer of Saccharothrix tangerinus Kinoshita et al. as Umezawaea tangerina gen. nov., comb. nov. Int J Syst Evol Microbiol 57:2758–2761. https://doi.org/10.1099/ijs.0.64985-0
Hoshino S, Wong CP, Ozeki M, Zhang H, Hayashi F, Awakawa T, Asamizu S, Onaka H, Abe I (2018) Umezawamides, new bioactive polycyclic tetramate macrolactams isolated from a combined-culture of Umezawaea sp. and mycolic acid-containing bacterium. J Antibiot 71:653–657. https://doi.org/10.1038/s41429-018-0040-4
Hoshino S, Ozeki M, Awakawa T, Morita H, Onaka H, Abe I (2018) Catenulobactins A and B, heterocyclic peptides from culturing Catenuloplanes sp. with a mycolic acid-containing bacterium. J Nat Prod 81:2106–2110. https://doi.org/10.1021/acs.jnatprod.8b00261
Shapiro JA, Wencewicz TA (2016) Acinetobactin isomerization enables adaptive iron acquisition in Acinetobacter baumannii through pH-triggered siderophore swapping. ACS Infect Dis 2:157–168. https://doi.org/10.1021/acsinfecdis.5b00145
Hoshino S, Okada M, Awakawa T, Asamizu S, Onaka H, Abe I (2017) Mycolic acid containing bacterium stimulates tandem cyclization of polyene macrolactam in a lake sediment derived rare actinomycete. Org Lett 19:4992–4995. https://doi.org/10.1021/acs.orglett.7b02508
Skellam EJ, Stewart AK, Strangman WK, Wright JL (2013) Identification of micromonolactam, a new polyene macrocyclic lactam from two marine Micromonospora strains using chemical and molecular methods: clarification of the biosynthetic pathway from a glutamate starter unit. J Antibiot 66:431–441. https://doi.org/10.1038/ja.2013.34
Arai MA, Tanaka M, Tanouchi K, Ishikawa N, Ahmed F, Sadhu SK, Ishibashi M (2017) Hes1-binding compounds isolated by target protein oriented natural products isolation (TPO-NAPI). J Nat Prod 80:538–543. https://doi.org/10.1021/acs.jnatprod.6b01072
Oh DC, Poulsen M, Currie CR, Clardy J (2011) Sceliphrolactam, a polyene macrocyclic lactam from a wasp-associated Streptomyces sp. Org Lett 13:752–755. https://doi.org/10.1021/ol102991d
Derewacz DK, Covington BC, McLean JA, Bachmann BO (2015) Mapping microbial response metabolomes for induced natural product discovery. ACS Chem Biol 10:1998–2006. https://doi.org/10.1021/acschembio.5b00001
Schulze CJ, Donia MS, Siqueira-Neto JL, Ray D, Raskatov JA, Green RE, McKerrow JH, Fischbach MA, Linington RG (2015) Genome-directed lead discovery: biosynthesis, structure elucidation, and biological evaluation of two families of polyene macrolactams against Trypanosoma brucei. ACS Chem Biol 10:2373–2381. https://doi.org/10.1021/acschembio.5b00308
Hoshino S, Ozeki M, Wong CP, Zhang H, Hayashi F, Awakawa T, Morita H, Onaka H, Abe I (2018) Mirilactams C-E, novel polycyclic macrolactams isolated from combined-culture of Actinosynnema mirum NBRC 14064 and mycolic acid-containing bacterium. Chem Pharm Bull 66:660–667. https://doi.org/10.1248/cpb.c18-00143
Raju R, Piggott AM, Conte MM, Capon RJ (2010) Heronamides A-C, new polyketide macrolactams from an Australian marine-derived Streptomyces sp. A biosynthetic case for synchronized tandem electrocyclization. Org Biomol Chem 8:4682–4689. https://doi.org/10.1039/c0ob00267d
Sugiyama R, Nishimura S, Kakeya H (2013) Stereochemical reassignment of heronamide A, a polyketide macrolactam from Streptomyces sp. Tetrahedron Lett 54:1531–1533. https://doi.org/10.1016/j.tetlet.2013.01.012
Zhang W, Li S, Zhu Y, Chen Y, Chen Y, Zhang H, Zhang G, Tian X, Pan Y, Zhang S, Zhang W, Zhang C (2014) Heronamides D-F, polyketide macrolactams from the deep-sea-derived Streptomyces sp. SCSIO 03032. J Nat Prod 77:388–391. https://doi.org/10.1021/np400665a
Sugiyama R, Nishimura S, Matsumori N, Tsunematsu Y, Hattori A, Kakeya H (2014) Structure and biological activity of 8-deoxyheronamide C from a marine-derived Streptomyces sp.: heronamides target saturated hydrocarbon chains in lipid membranes. J Am Chem Soc 136:5209–5212. https://doi.org/10.1021/ja500128u
Ben Fekih I, Zhang C, Li YP, Zhao Y, Alwathnani HA, Saquib Q, Rensing C, Cervantes C (2018) Distribution of arsenic resistance genes in prokaryotes. Front Microbiol 9:2473. https://doi.org/10.3389/fmicb.2018.02473
Chen J, Rosen BP (2020) The arsenic methylation cycle: how microbial communities adapted methylarsenicals for use as weapons in the continuing war for dominance. Front Environ Sci 8:43. https://doi.org/10.3389/fenvs.2020.00043
Kuramata M, Sakakibara F, Kataoka R, Abe T, Asano M, Baba K, Takagi K, Ishikawa S (2015) Arsenic biotransformation by Streptomyces sp. isolated from rice rhizosphere. Environ Microbiol 17:1897–1909. https://doi.org/10.1111/1462-2920.12572
Miyashita S, Fujiwara S, Tsuzuki M, Kaise T (2012) Cyanobacteria produce arsenosugars. Environ Chem 9:474–484. https://doi.org/10.1071/en12061
Xue XM, Raber G, Foster S, Chen SC, Francesconi KA, Zhu YG (2014) Biosynthesis of arsenolipids by the cyanobacterium Synechocystis sp. PCC 6803. Environ Chem 11:506–513. https://doi.org/10.1071/en14069
Kuramata M, Sakakibara F, Kataoka R, Yamazaki K, Baba K, Ishizaka M, Hiradate S, Kamo T, Ishikawa S (2016) Arsinothricin, a novel organoarsenic species produced by a rice rhizosphere bacterium. Environ Chem 13:723–731. https://doi.org/10.1071/en14247
Nadar VS, Chen J, Dheeman DS, Galvan AE, Yoshinaga-Sakurai K, Kandavelu P, Sankaran B, Kuramata M, Ishikawa S, Rosen BP, Yoshinaga M (2019) Arsinothricin, an arsenic-containing non-proteinogenic amino acid analog of glutamate, is a broad-spectrum antibiotic. Commun Biol 2:131. https://doi.org/10.1038/s42003-019-0365-y
Cheng J, Ji W, Ma S, Ji X, Deng Z, Ding W, Zhang Q (2021) Characterization and mechanistic study of the radical SAM enzyme ArsS involved in arsenosugar biosynthesis. Angew Chem Int Ed Engl 60:7570–7575. https://doi.org/10.1002/anie.202015177
Yao Y, He J, Chen F, Dong M (2024) Arsinothricin biosynthesis involving a radical SAM enzyme for noncanonical SAM cleavage and C-As bond formation. J Am Chem Soc 146:21214–21219. https://doi.org/10.1021/jacs.4c06403
Cruz-Morales P, Kopp JF, Martinez-Guerrero C, Yanez-Guerra LA, Selem-Mojica N, Ramos-Aboites H, Feldmann J, Barona-Gomez F (2016) Phylogenomic analysis of natural products biosynthetic gene clusters allows discovery of arseno-organic metabolites in model Streptomycetes. Genome Biol Evol 8:1906–1916. https://doi.org/10.1093/gbe/evw125
Hoshino S, Ijichi S, Asamizu S, Onaka H (2023) Insights into arsenic secondary metabolism in actinomycetes from the structure and biosynthesis of bisenarsan. J Am Chem Soc 145:17863–17871. https://doi.org/10.1021/jacs.3c04978
Wang B, Guo F, Huang C, Zhao H (2020) Unraveling the iterative type I polyketide synthases hidden in Streptomyces. Proc Natl Acad Sci USA 117:8449–8454. https://doi.org/10.1073/pnas.1917664117
Jiang Y, Matsumoto T, Kuranaga T, Lu S, Wang W, Onaka H, Kakeya H (2021) Longicatenamides A-D, two diastereomeric pairs of cyclic hexapeptides produced by combined-culture of Streptomyces sp. KUSC_F05 and Tsukamurella pulmonis TP-B0596. J Antibiot 74:307–316. https://doi.org/10.1038/s41429-020-00400-3
Pan C, Kuranaga T, Cao X, Suzuki T, Dohmae N, Shinzato N, Onaka H, Kakeya H (2021) Amycolapeptins A and B, cyclic nonadepsipeptides produced by combined-culture of Amycolatopsis sp. and Tsukamurella pulmonis. J Org Chem 86:1843–1849. https://doi.org/10.1021/acs.joc.0c02660
Asamizu S, Pramana AAC, Kawai SJ, Arakawa Y, Onaka H (2022) Comparative metabolomics reveals a bifunctional antibacterial conjugate from combined-culture of Streptomyces hygroscopicus HOK021 and Tsukamurella pulmonis TP-B0596. ACS Chem Biol 17:2664–2672. https://doi.org/10.1021/acschembio.2c00585
Pan C, Ikeda H, Minote M, Tokuda T, Kuranaga T, Taniguchi T, Shinzato N, Onaka H, Kakeya H (2024) Amoxetamide A, a new anoikis inducer, produced by combined-culture of Amycolatopsis sp. and Tsukamurella pulmonis. J Antibiot 77:66–70. https://doi.org/10.1038/s41429-023-00668-1
Acknowledgements
The projects described in this review were conducted at the Laboratory of Natural Products Chemistry, Graduate School of Pharmaceutical Sciences, The University of Tokyo (with Prof. Ikuro Abe as the principal investigator) and the Laboratory of Microbial Science, Department of Life Science, Faculty of Science, Gakushuin University (with Prof. Hiroyasu Onaka as the principal investigator, formerly the Laboratory of Microbial Enzyme Potential endowed by Amano Enzyme Inc, Graduate School of Agricultural and Life Sciences, The University of Tokyo). I would like to express my sincere gratitude to Prof. Ikuro Abe, Prof. Hiroyasu Onaka, Prof. Toshiyuki Wakimoto (Hokkaido University), Prof. Hiroyuki Morita (Toyama University), Prof. Takayoshi Awakawa (Riken), Prof. Shumpei Asamizu (Kobe University), Prof. Masahiro Okada (Kanagawa University), Prof. Takahiro Mori (The University of Tokyo), Prof. Yudai Matsuda (City University of Hong Kong), and all the members who contributed to our research conducted in the both laboratories. Finally, I sincerely appreciate the Japanese Society of Pharmacognosy (JSP) for selecting me for the Award in 2024 and providing me the opportunity to deliver the award lecture at the 70th Annual Meeting of the JSP.
Funding
This work was supported by JSPS KAKENHI (Grant Numbers JP23H04543, JP22K20566, JP18H02120, JP16H06443, JP16K13084 and JP15H01836), JST/NSFC Strategic International Collaborative Research program Japan–China “Exploitation of the cryptic secondary metabolites from plant microbiome through biological interaction”, JSPS Research Fellowships for Young Scientists (Grant Number JP16J01117), and a grant-in-aid from the Amano Enzyme Foundation.
Author information
Authors and Affiliations
Contributions
SH prepared this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to a retrospective open access order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Hoshino, S. Exploring new natural products by utilizing untapped secondary metabolic pathways in actinomycetes. J Nat Med 79, 465–476 (2025). https://doi.org/10.1007/s11418-025-01903-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1007/s11418-025-01903-9