

Abstract
Tauopathies are a class of neurodegenerative diseases, including Alzheimer’s disease, frontotemporal dementia and progressive supranuclear palsy, that are associated with the pathological aggregation of tau protein in neurofibrillary tangles (NFT). Studies have characterized tau as a “prion-like” protein given its ability to form distinct, stable amyloid conformations capable of transcellular and multigenerational propagation in clonal fashion. It has been proposed that progression of tauopathy could be due to the prion-like propagation of tau, suggesting the possibility that end-stage pathologies like NFT formation may require an instigating event such as tau seeding. To investigate this, we applied a novel human induced pluripotent stem cell (hiPSC) system we have developed to serve as a human neuronal model. We introduced the tau repeat domain (tau-RD) with P301L and V337M (tau-RD-LM) mutations into hiPSC-derived neurons and observed expression of tau-RD at levels similar to total tau in postmortem AD brains. Tau aggregation occurred without the addition of recombinant tau fibrils. The conditioned media from tau-RD cultures contained tau-RD seeds, which were capable of inducing aggregate formation in homotypic mode in non-transduced recipient neuronal cultures. The resultant NFTs were thioflavin-positive, silver stain-positive, and assumed fibrillary appearance on transmission electron microscopy (TEM) with immunogold, which revealed paired helical filament 1 (PHF1)-positive NFTs, representing possible recruitment of endogenous tau in the aggregates. Functionally, expression of tau-RD caused neurotoxicity that manifested as axon retraction, synaptic density reduction, and enlargement of lysosomes. The results of our hiPSC study were reinforced by the observation that Tau-RD-LM is excreted in exosomes, which mediated the transfer of human tau to wild-type mouse neurons in vivo. Our hiPSC human neuronal system provides a model for further studies of tau aggregation and pathology, as well as a means to study transcellular propagation and related neurodegenerative mechanisms.
Keywords: tau, neurofibrillary tangles, induced pluripotent stem cells, exosomes, propagation
1. Introduction
Tau pathology is characterized by the presence of abnormally hyperphosphorylated tau protein that has misfolded into insoluble aggregates known as neurofibrillary tangles (NFT). Tau protein is abundant in neurons, acting to stabilize microtubules and facilitate axonal transport. When hyperphosphorylated, tau has a heightened tendency to misfold, losing its ability to interact with microtubules (Iqbal et al., 2009). An accumulation of pathologically aggregated tau protein is often observed in postmortem brains from patients suffering from tauopathies. In Alzheimer’s disease (AD), there is build-up of both amyloid plaques and neurofibrillary tangles, with the appearance of NFTs correlated with clinical expression of dementia (Alafuzoff et al., 1987; Arriagada et al., 1992; Knopman et al., 2013).
The processing, misfolding and aggregation of tau are all implicated in the progression of neurodegeneration. Abnormal hyperphosphorylation is an early pathological event that precedes successive truncations at both the N- and C-termini, leaving the tau repeat domain (tau-RD) at the core of the cleaved protein (Garcia-Sierra et al., 2003; Mondragon-Rodriguez et al., 2008). The repeat domain of tau, which interacts with microtubules, aggregates more readily, likely due to tau-RD’s propensity to form β – pleated sheets and assume amyloid-patterned aggregates (Mandelkow et al., 2007). Tau-RD has been found to be a primary component of NFTs (Endoh et al., 1993; Novak et al., 1991).
Patient (human) derived induced pluripotent stem cells (hiPSC) have emerged as a powerful new tool to model human neurodegenerative diseases (Yuan and Shaner, 2013). Human neuronal cultures are grown from hiPSC derived from cells obtained through skin biopsies of carriers of tau mutations associated with frontotemporal dementia (FTD). Previous human models of tauopathy have demonstrated early pathological phenotypes, such as hyperphosphorylated tau and pre-tangle AT-8 positive puncta, yet failed to manifest NFT formation (Fong et al., 2013; Iovino et al., 2015). NFTs were also absent following overexpression of full-length 4-repeat (4R) tau (Mertens et al., 2013). Tau aggregation, as demonstrated on Western blot, can be induced with exogenous tau fibrils in hiPSCs overexpressing full-length tau; albeit, the resultant NFTs do not exhibit thioflavin staining, a confirmation of β -pleated sheet formation (Medda et al., 2016; Verheyen et al., 2015). However, overexpression of both the presenilin-1 (PS-1) familial mutations and the amyloid precursor protein (APP) have been shown to initiate hyperphosphorylation of tau as well as formation of SDS-insoluble, silver stain-positive NFTs in 3-dimensional (3-D) cultures (Choi et al., 2014).
Tau-RD has been shown to spread from cell-to-cell (Frost et al., 2009), and has been demonstrated to form distinct aggregate species, which can be transmitted to other cells, like prions (Sanders et al., 2014). It has further been demonstrated that aggregates found in human brain tissue from patients with tauopathies, can induce new aggregates in a naïve mouse brain (Clavaguera et al., 2013; Sanders et al., 2014). Recently, we demonstrated that neuronally-derived exosomes (NDEs) from AD patients seeded tau aggregation and induced AD-like neuropathology in normal mouse brains (Winston et al., 2016). Exosomes represent a subclass of secreted membrane vesicles that have been shown to shuttle protein cargo and mRNAs (Valadi et al., 2007) for intracellular communication between cells, to eliminate damaged or excess protein cargo (Raposo and Stoorvogel, 2013), and to mediate the propagation of Aβ peptides (Rajendran et al., 2006) and prion proteins (Fevrier et al., 2004).
We hypothesized that end-stage pathologies, like NFT formation, require an instigating event such as prion-like seeding, which may be lacking in previous human neuronal cell culture systems. We observed that Tau-RD is excreted from the neurons inside exosomes, which mediated the transfer of tau to naïve mouse neurons in vivo. We then demonstrated the intercellular transfer of tau in a novel model of human tauopathy. In our model, expression of tau-RD was sufficient to initiate NFT production, with continued spread of tau pathology propagating between neurons via the intracellular media. This hiPSC system can serve as a human neuronal model for studying tauopathy, neurodegeneration, and the mechanisms of pathological seed propagation both in vitro and in vivo.
2. Material and Methods
2.1 Generation of neuronal cultures
Neuronal cultures derived from non-demented controls (NDC) and PS-1 A246E mutation carriers (causative of early onset familial AD) were generated per a previously published protocol (Yuan et al., 2011). Neural stem cells (NSC) were seeded at a density of 150,000 cells/cm2 on either Matrigel-coated [70 μ g/mL] plastic cell culture dishes or onto glass coverslips coated with both polyornithine [20 μ g/mL] and Matrigel. NSCs were grown to 80% confluency, at which time neuronal differentiation was initiated through withdrawal of fibroblast growth factor (bFGF) from the NSC media (DMEM-12, 1% N-2, 2% B-27, Pen-Strep, 20 ng/mL bFGF).
2.2 Preparation of the soluble and insoluble portion of the protein
We followed the previously published protocol (Higuchi et al., 2002; Rissman et al., 2004) with adaptation for cell culture. NDC-derived cells were collected directly from confluent 6-well plates and subjected to sequential extraction with reassembly buffer (RAB), radioimmunoprecipitation assay buffer (RIPA), and formic acid. Collected cells were re-suspended in RAB [100 mM MES, 1 mM EGTA, 0.5 mM MgSO4, 0.75 mM NaCl, protease inhibitor cocktail set 1 (Calbiochem), phosphatase inhibitor single-use cocktail (Thermo Fisher)] and incubated on ice for 30 minutes. Samples were then centrifuged at 40,000 × g for 40 minutes at 4°C. Supernatant was collected and the pellet was re-suspended with RIPA buffer [50mM Tris pH 7.4, 150 mM NaCl, Triton-100x, 1 mM EDTA pH 7.5, 0.1% SDS, 0.25% Na-Deoxycholate, 1 mM EGTA, protease inhibitor cocktail set 1 (Calbiochem, #539131), phosphatase inhibitor single-use cocktail (Thermo Fisher, #78428)] and centrifuged at 40,000 × g for 20 minutes at 4°C. Supernatant was again collected and the cell pellet was re-suspended in 50 μl of 70% formic acid and sonicated 3 times (10 seconds/sonication), prior to centrifugation at 135,000 × g for 1 hour at 4°C. The sample was diluted 1:20 in formic acid neutralization buffer [1 M Tris, 0.5 M NaH2PO4] and its concentration determined by bicinchoninic acid protein assay (BCA).
2.3 Western blot
10–20 μg of soluble or insoluble protein was collected as previously described, diluted with 4x Laemmli sample buffer (BioRad), and resolved on 12% SDS polyacrylamide gel. Protein was transferred to polyvinylidene difluoride (PVDF) membranes using Trans-Blot SD Semi-Dry transfer cell (BioRad). Blots were blocked in 5% non-fat milk in tris-buffered saline (TBS) with 0.1% Tween-20 (TBST) at room temperature for 45 minutes on a platform shaker. Primary antibodies were applied overnight at 4°C in blocking solution. The primary antibodies used were paired helical filament 1 (PHF1) (1:2000; gift from Peter Davies), tau-RD rabbit anti-tau antibody (1:2000; Abcam ab64193), total tau-K9JA (1:30,000; DAKO), CP13 (1: 200; gift from Peter Davies), Alz50 (1:250; gift from Peter Davies), β -III-tubulin (1:4000; Covance), β -actin (1:8000; Sigma). Following a series of washes, blots were incubated with horseradish peroxidase (HRP) conjugated secondary antibodies (1:2000; Thermo-Fisher). Enhanced chemiluminescence (ECL) substrate was applied before visualization by GelDoc (BioRad). Blot analysis was performed on ImageLab 5.2.1 (BioRad). Graphs were created using Prism (GraphPad).
2.4 Immunofluorescence studies
NDC and PS-1-derived cultures were fixed at room temperature with 4% paraformaldehyde (Lamp2 samples were post-treated with cold 100% methanol for 5 minutes at −20°C) 4 days post-transduction or transfection. Fixed cultures were blocked for 45 minutes at room temperature in blocking solution (3% bovine serum albumin (BSA), 0.3% Triton X-100, 1x phosphate buffered saline (PBS)), stained for 2 hours at room temperature or overnight at 4°C, and washed twice with PBS (5 minutes/wash). The primary antibodies used were Map2b (1:500; Sigma), β -III-tubulin (1:500; Covance), synapsin (1:2000; Abcam), lamp2 (1:500; DSHB). All secondary antibodies were diluted 1:1000 in blocking solution and applied for 45 minutes at room temperature. Antifade solution (Prolong, Thermo) was applied prior to mounting the coverslips to glass slides, in an attempt to preserve the immunofluorescent signal. Samples were imaged with a Leica confocal microscope and data collection and analysis was performed using ImageJ.
2.5 Thioflavin staining
NDC-derived four-week-differentiated neurons, grown on glass coverslips in 24-well plates, were fixed in 4% paraformaldehyde for 20 minutes at room temperature. Cells were stained with primary and secondary antibodies according to standard protocols. Following staining, cells were washed for 10 minutes in H2O and incubated in 300 μl of 0.015% Thioflavin-S (Sigma) in 50% ethanol for 10 minutes. The cells were then washed twice (4 minutes/wash) in 50% ethanol, followed by one 4-minute wash in 30% ethanol, and 2 additional washes (5 minutes/wash) in 30% ethanol. A final rinse with H2O was performed prior to mounting on a slide with Prolong antifade solution.
2.6 Gallyas Silver Stain
NDC-derived four-week-differentiated neurons, grown on glass coverslips in 24-well plates, were fixed with 4% PFA according to the previously described protocol (Gallyas, 1971). Following fixation, a 5-minute wash in 5% periodic acid and two 5-minute washes in double de-ionized water (DDW) were performed. Samples were then placed in alkalic silver iodide-solution for 1 minute, followed by a 10-minute wash in 0.5% acetic acid. During development in a solution of 5% sodium bicarbonate, 0.2% ammonium nitrate, 0.2% silver nitrate, 1% tungstosilicic acid and 9.25% PFA, samples were checked under a light microscope for the presence of brown densities. One 3-minute wash in 0.5% acetic acid were performed in order to terminate the reaction. Residual acetic acid was removed by washing with DDW for 5 minutes. Samples were then placed in a 0.1% gold chloride solution for 5 minutes, followed by a rinse in DDW, and incubation in DDW for an additional 5 minutes. A 10-minute incubation in 1% sodium thiosulfate solution ensued, followed by a 10-minute wash in DDW before sealing with ProLong-Gold antifade onto a glass slide. All solutions are % weight/volume.
2.7 Neurite length studies
NDC- and PS1- derived four-week-differentiated neuronal cultures, plated on 12 mm glass coverslips, were washed with PBS and incubated in opti-MEM for 30 minutes before transiently transfecting with 2 μg tau-PP-YFP + 1 μg mCherry, tau-WT-YFP + 1 μg mCherry, tau-LM-YFP + 1 μg mCherry or 1 μg mCherry using Lipofectamine 2000 transfection reagent (1:2; DNA:Lipofectamine, Thermo Fisher). Cells were incubated in DNA/Lipofectamine/opti-MEM for 2 hours and washed with 500 μl PBS. Fresh media was added and the cells were incubated until fixation at 2 or 4 days post-transfection. Prior to fixation, live imaging was performed on days 1, 2, and 4 using a Leica upright fluorescent microscope. Fixed cells were imaged using a confocal microscope. β -III-tubulin (1:500; Covance) was used to label all neurites and Map2b (1:500; Sigma) was used to distinguish dendrites. Axons were defined as β -III-tubulin+/Map2b-; dendrites β -III-tubulin+/Map2b+. Neurite lengths were measured using the NeuronJ plugin for ImageJ. Statistics were performed and figures were created using Prism.
2.8 Lamp2/synapsin analysis
NDC- and PS1- derived four-week-differentiated neuronal cultures, were transfected with YFP or tau-LM-YFP following the instructions from the manufacturer of Lipofectamine 2000. Two days after transfection, cells were fixed and stained according to the immunofluorescence procedure. The Lamp2 staining included a post-fix in 100% EtOH for 5 minutes at −20°C. Images were taken as stacks on a Leica confocal microscope and analyzed using ImageJ. Synaptic density was performed by counting the number of synapses, which co-localize with the dendrites of the transfected neuron. Lysosomal volume was calculated by rendering a 3D image of the stack and assigning a volume to each pixel to calculate the total lysosomal volume/cell. The antibodies used included anti-Lamp2 (1:500), anti- β -III-tubulin (TujI 1:500), anti-synapsin (1:2000) and anti-Map2b (1:500).
2.9 Generation of lentivirus
We followed the lentivirus production protocol (Sanders et al., 2014). Briefly, HEK293T cells were transfected in 3 × 15 cm tissue culture plates with lentiviral constructs using the calcium phosphate method. Media samples were collected on 2 and 3 days post-transfection. The media was centrifuged at 1,000 RPM for 5 minutes and filtered using a 0.2 μm filter, in order to remove cellular particles. The filtered media were concentrated by centrifugation at 50,000 × g for 2 hours at room temperature and resuspended in neuronal culture media at a volume equal to 1/100th of the total collected volume. The lentivirus titer was measured by transducing HEK293T cells at varying concentrations and performing FACS analysis in transducible units (TU)/mL.
2.10 Generation of tau-RD-LM seeds conditioned media
NDC-derived four-week-differentiated neuronal cultures, grown in 6-well plates, were transduced at 1.2×106 TU with tau-LM-YFP and incubated for 24 hours. The following morning, the media was aspirated, cells were washed with PBS, and fresh media was applied. To ensure removal of virus, cells were washed with PBS multiple times and given fresh media for 2 additional days. Three days post-transduction, 2 mL of fresh neuronal media was added to each well and the cells incubated for 3 additional days. Conditioned media was collected and centrifuged at 1000 RPM for 5 minutes at 4°C, in order to clear cell debris.
2.11 Transfection
NDC- and PS1-derived four-week-differentiated neuronal cultures, grown on 12mm glass coverslips, were transfected with 2 μg YFP or 2 μg tau-RD-YFP constructs with Lipofectamine 2000 1:2 (DNA:Lipofectamine). Transfection of each line was performed in triplicate.
2.12 Lentiviral transduction
NDC-derived four-week-differentiated neuronal cultures were transduced with lentivirus at low titer (1.7×105 transducible units/cell), or high titer (3.4×105 transducible units/cell).
2.13 Transmission Electron Microscopy
NDC-derived four-week-differentiated neurons grown on Mattek dishes were transfected with Lipofectamine 2000 (Thermo Fisher) per manufacturer protocol. The neurons were fixed with EM-grade 4% paraformaldehyde, embedded, and sectioned with an ultramicrotome. Electron micrographs were obtained at a magnification of × 25,000 using a Zeiss OM 10 electron microscope (Masliah et al., 1996).
2.14 Exosome preparation
Exosomes were isolated from conditioned media collected from human neuronal culture differentiated from iPSC. Two mL of conditioned media were incubated with 0.4 mL ExoQuik-TC precipitation solution, rotating overnight at 4°C. After centrifugation at 1500 × g for 1 hour at 4°C, supernatant was collected and the resultant pellet was resuspended in 300 μ L of 1x PBS with protease and phosphatase inhibitor cocktail EDTA-free then stored at −80°C.
2.15 Characterization of exosomes
Nanoparticle tracking analysis (NTA) was used to characterize neuronally differentiated, induced pluripotent stem cell – derived exosomes (NiPSCEs) based on size distribution. Three replicates of NiPSCE suspensions were pooled in 100 μl of 1X PBS, diluted 1:300 and visualized with a NanoSight LM10 instrument as described (Mitsuhashi et al., 2013). Protein concentrations for NiPSCE suspensions were determined using a BCA Protein Assay kit (Pierce Biotechnology). 5 μ g of protein was loaded and electrophoretically separated on a Mini-PROTEAN TGX Precast Gels (Bio-Rad Laboratories, Hercules, California). Proteins were transferred to 0.2 μ m PVDF membrane and incubated in primary antibody followed by HRP-linked secondary antibodies and developed with a chemiluminescence western blot detection kit (Pierce Super Signal). For immunoblots, rabbit anti-human tau, K9JA (1: 32,000; DAKO) was used to detect human total tau. Mouse anti-phospho tau, PHF1 (S396/404; 1:500; gift from Dr. P. Davies, Albert Einstein, NY) and mouse anti-phospho tau, AT8 (S202/T205; 1:500; Pierce Biotechnology) was used to detect phosphorylated tau (P-tau). Exosome surface marker, Anti-CD63 antibody biotin [MEM-259] (1:1000, ABCAM) was used as a loading control.
2.16 Injection of NiPSCEs
For all in vivo experiments, 2 μ L of NiPSCE suspension isolated from cell culture media was injected into the right hippocampus of wild-type, female, C57/BL6 mice (n = 5/group, 3 – 4 month old) at the level of (−2.0, +1.5, −1.3) (from Bregma, lateral, into) and analyzed 1 month (1m) post-injection using immunohistochemistry (IHC). Control mice were injected with 1X PBS. Brain slices containing hippocampus were probed using primary and secondary antibodies as previously described.
Thirty μ m-thick frozen sections were cut on a sliding microtome and stored at −20°C in cryoprotectant solution (20% glycerol and 30% ethylene glycol in 0.1 m phosphate buffer). For immunostaining, K9JA (1:10,000) was used to detect htau in free-floating sections containing hippocampus. Endogenous peroxidase was quenched with 0.3% hydrogen peroxide to reduce free aldehydes. Reaction product was developed using a nickel-enhanced glucose oxidase method.
2.17 Statistical analysis
Student’s t-test and ANOVA with post-test analysis were performed using GraphPad Prism.
3. Results
3.1 Tau aggregate formation in human neurons
We took advantage of the aggregation propensity of the tau repeat domain to test if tau-RD would drive the formation of aggregates and NFTs in a hiPSC model. The hiPSC lines used were previously described and fully characterized (Table 1) (Israel et al., 2012; Liu et al., 2014). Four-week-differentiated human neuronal cultures derived from NDC were transduced with lentivirus carrying yellow fluorescent protein (YFP), tau-RD-WT-YFP, tau-RD-PP-YFP, or tau-RD-LM-YFP (Fig. 1A). We have previously shown that these neurons generate voltage-dependent action potentials and electric currents, and possess spontaneous synaptic activity, which could be reversibly blocked by a GABAA receptor antagonist (Israel et al., 2012). We have also performed a time-course, which demonstrated that tau protein expression is stable after the cultures have been differentiated for four weeks (Supplementary Fig. 1A, B). qPCR was used to characterize the expressed tau isoforms and revealed primarily 3R tau in the iPSC derived human neurons (Supplementary Fig. 1C).
Table 1.
Clinical information regarding the hiPSC lines used
Fig. 1.
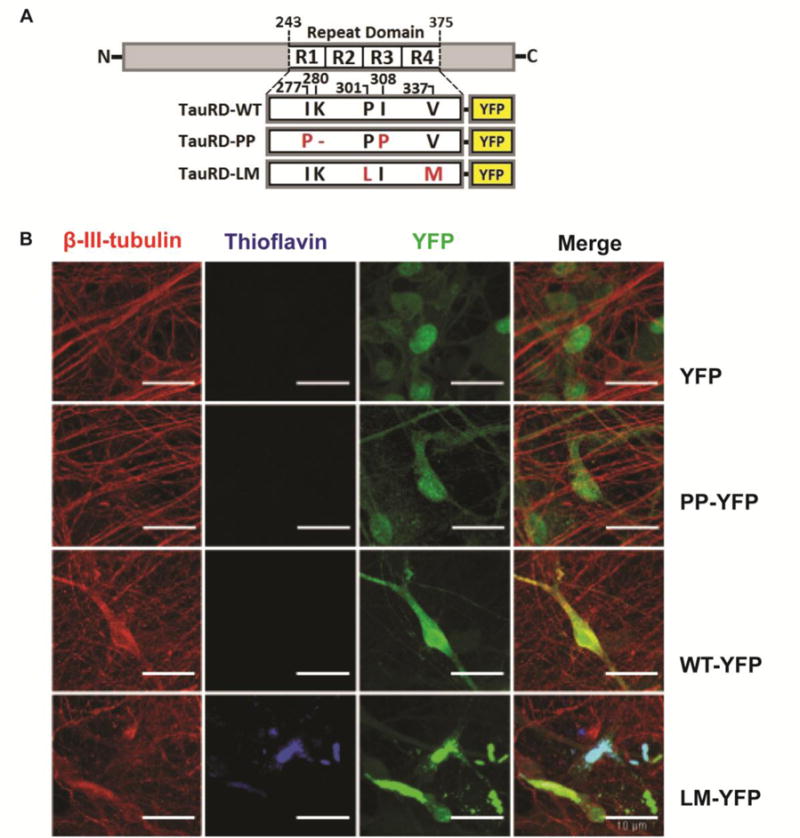
Neurofibrillary tangle formation in neurons derived from hiPSC. (A) Schematic of tau-RD constructs. Tau-RD-WT-YFP contains the wild type repeat domain. Tau-RD-PP-YFP contains the repeat domain with two substitutions of isoleucine-to-proline at positions 277 and 308. Tau-RD-LM-YFP contains the repeat domain with two mutations, both cause familial frontotemporal dementia with parkinsonism (FTLD-17). The tau-RD is tagged with YFP for visualization. (B) Neuronal cultures derived from NDC hiPSC, which were transduced with tau-RD lentivirus, show NFTs, which are thioflavin positive, only in cultures transduced with tau-RD-LM (arrows). Scale bar is 10μm
Tau-RD-WT-YFP contains the wild-type repeat domain. Tau-RD-PP-YFP is an anti-aggregator whose repeat domain contains two isoleucine-to-proline mutations. These mutations prevent β -pleated sheet formation, and this construct serves as a control to differentiate whether the observed effect is due to over-expression of soluble tau-RD or to aggregation of tau-RD (Khlistunova et al., 2006; Khlistunova et al., 2007). In contrast, tau-RD-LM-YFP is a super-aggregator whose repeat domain contains two missense mutations, P301L and V337M, which have individually been identified as causative of frontotemporal dementia with parkinsonism-17 (FTDP-17).
Expression of tau-RD-LM has been shown to cause tau aggregation (Frost et al., 2009; Kfoury et al., 2012). Transduction with YFP alone served as a control for the viral transduction itself. Compared to YFP alone, we observed, on average, one YFP-positive aggregate per 10x viewing field in cultures transduced with tau-RD-LM-YFP (Fig. 1B). The aggregates formed spontaneously, without exogenous induction with fibrils. Tau-RD-PP-YFP did not induce aggregate formation, consistent with the hypothesis that aggregation is dependent on β -pleated sheet formation. There were also no visible aggregates in the tau-RD-WT-YFP culture (Fig. 1B). The aggregates were recognized by an antibody specific to the tau repeat domain (Abcam) (Fig. 2B). The aggregates were also partially stained by Alz-50, an antibody that recognizes misfolded tau (Fig. 2C) (Jicha et al., 1997). Alz-50 does not recognize tau-RD and thus, its recognition suggests that endogenous tau is recruited by tau-RD and assumes a misfolded conformation.
Fig. 2.

Aggregated tau is SDS insoluble. (A) Western blot of NDC-derived four-week human neuronal cultures transduced with YFP, tau-RD-PP-YFP, tau-RD-WT-YFP, tau-RD-LM-YFP, shows aggregated tau, which is SDS insoluble. This process appears to be non-phosphorylation dependent based on analysis of PHF1 and CP13 staining. (B) NDC-derived four-week human neuronal cultures transduced with tau-RD-LM-YFP form aggregates, which are positive for the antibody tau-RD, which recognizes the repeat domain. (C) NDC-derived four-week human neuronal cultures transduced with tau-RD-LM-YFP form aggregates, which are positive for the antibody Alz50, which recognizes conformational changes in tau, suggesting recruitment of endogenous tau into the aggregates.
3.2 Tau aggregates are detergent insoluble
Four-week neuronal cultures derived from NDC were transduced with lentivirus containing YFP, tau-RD-PP-YFP, tau-RD-WT-YFP and tau-RD-LM-YFP. We have observed that aggregate formation is dependent on the viral titer. At low viral titer (1.7×105 transducible units), minimal or no aggregates formed, while at a high titer (3.4×105 transducible units), higher numbers of aggregates were formed. Aggregation occurred spontaneously 4–7 days after viral induction. At high titer, where spontaneous NFT formation was observed, expression of tau-RD was nine times wild-type tau levels; however, this level of overexpression is comparable with the pathological level of tau detected in human AD brains (Khatoon et al., 1992; Khatoon et al., 1994).
Both tau-RD-WT-YFP and tau-RD-LM-YFP formed tau aggregates that were found in the SDS insoluble fraction (Fig. 2A), whereas YFP and tau-RD-PP-YFP did not (Fig. 2A). Formation of tau aggregates within tau-RD-LM cultures is consistent with previous studies, which have shown that FTD mutations increase the aggregation of tau (Alonso Adel et al., 2004; Hutton et al., 1998). Lack of aggregation within tau-RD-PP-YFP cultures is consistent with studies showing that tau aggregation is promoted by β-pleated sheet formation (Mandelkow et al., 2007). Furthermore, tau aggregation within tau-RD-WT-YFP cultures demonstrates that the repeat domain is sufficient to induce aggregate formation, albeit it not as efficiently as tau-RD-LM-YFP (Fig. 2A). This tau-RD-dependent aggregation was independent of phosphorylation at the epitopes recognized by CP13 and PHF1, antibodies that recognize phosphorylation of tau at sites outside the repeat domain (Fig. 2A).
To determine if induction of tau aggregation by tau-RD-LM was specific to human neurons, we asked whether it could also occur in rat neurons. Embryonic (E18) rat cortical cultures were transduced with tau-RD-PP or tau-RD-LM, but no thioflavin-positive aggregates were observed using the same conditions, including the titer and incubation time, as for the human neurons (Fig. 3). To determine if simply more virus or incubation time was needed for the aggregate formation in rat neurons, the amount of virus was tripled and the neurons were cultured for an additional week, but once again, no aggregates formed. This finding suggests that human neuronal cultures are more efficient at generating tau aggregates and that tau aggregation may be a species-dependent event.
Fig. 3.

Rat primary cortical neurons do not exhibit thioflavin positive aggregates. Rat E18 cortical neuronal cultures were transduced with 3 times the high titer lentivirus carrying (A) tau-RD-PP-YFP or (B) tau-RD-LM-YFP on days in vitro (DIV) 3. Cultures did not show thioflavin positive puncta 10 days after transduction
3.3 Electron microscopy and silver stain evaluation of the tau aggregates
To further validate the identity of the NFTs, we performed transmission electron microscopy (TEM), which showed that fibrillar aggregates could be found both intracellularly and extracellularly. The fibrils were detected in NDC-derived human neuronal cultures transfected with tau-RD-LM, but not in cultures transfected with mCherry (Fig. 4A). The fibrils were positive for PHF1 on EM with immunogold (Fig. 4A), suggesting recruitment of endogenous tau as PHF1 recognizes phosphorylation of epitopes outside the repeat domain. Tau-RD-LM induced aggregates were also observed following Gallyas silver impregnation (Fig. 4B), a standard method for detecting NFTs in post-mortem human brains (Gallyas, 1971).
Fig. 4.

Tau-RD-LM induced aggregates are PHF1 immunoreactive by immunogold on EM and positive with silver stain. (A) Electron micrograph shows fibrils in tau-RD-LM transfected cultures, which are absent in the mCherry control. The fibrils are positive for the PHF1 (anti-phosphorylated tau) antibody on immunogold. Scale bar is 50 nm. (B) Silver staining of human neuronal cultures transduced with YFP, tau-RD-PP-YFP, tau-RD-WT-YFP or tau-RD-LM-YFP. Only tau-RD-LM-YFP stained positively with silver (scale bar 50μm)
3.4 Transcellular transfer of tau in human neurons
Previous reports have shown that recombinant tau fibrils can enter into cells by endocytosis (Frost et al., 2009; Holmes et al., 2014). However, it remains enigmatic whether tau seeds can enter into human neurons. To investigate this question, we tested whether conditioned media, from NDC-derived human neuronal cultures transduced with tau-RD-LM-YFP, contained seeds that might be capable of inducing aggregate formation in other cells. We first transduced neuronal cultures with YFP, tau-RD-PP-YFP, tau-RD-WT-YFP or tau-RD-LM-YFP under low titer conditions. Three days post-transduction, no tau aggregates were observed. This is consistent with previous observations that low titer conditions are insufficient to induce aggregate formation. Conditioned media was then collected from a separate culture that had been transduced with tau-RD-LM-YFP under high titer conditions and had previously demonstrated aggregation (Fig. 5A). The aforementioned neuronal cultures, YFP, tau-RD-PP-YFP, tau-RD-WT-YFP and tau-RD-LM-YFP, were incubated in this conditioned media. Three days after incubation, tau aggregates, which were identified as YFP-positive puncta, were observed in the cultures expressing tau-RD-LM-YFP (Fig. 5B, Supplementary Fig. 2). The amount of aggregation peaked between 7–10 days. Interestingly, on rare occasions, aggregates were found (approximately 2 aggregates per 113mm glass coverslip) in tau-RD-WT-YFP-expressing cultures incubated with conditioned media. These aggregates were observed at a significantly lower level than cultures transduced with tau-RD-LM-YFP, suggesting that homotypic templating is more efficient than heterotypic templating (Fig. 5B, Supplementary Fig. 2). The cultures transduced with tau-RD-PP-YFP did not form aggregates when incubated with conditioned media, further demonstrating that aggregation is dependent on β-pleated sheet formation. An untreated control culture of tau-RD-LM-YFP also lacked visible aggregates (Fig. 5B), indicating that aggregates and fibrils observed in the treated tau-RD-LM-YFP culture were induced by seeds originating from the conditioned media.
Fig. 5.
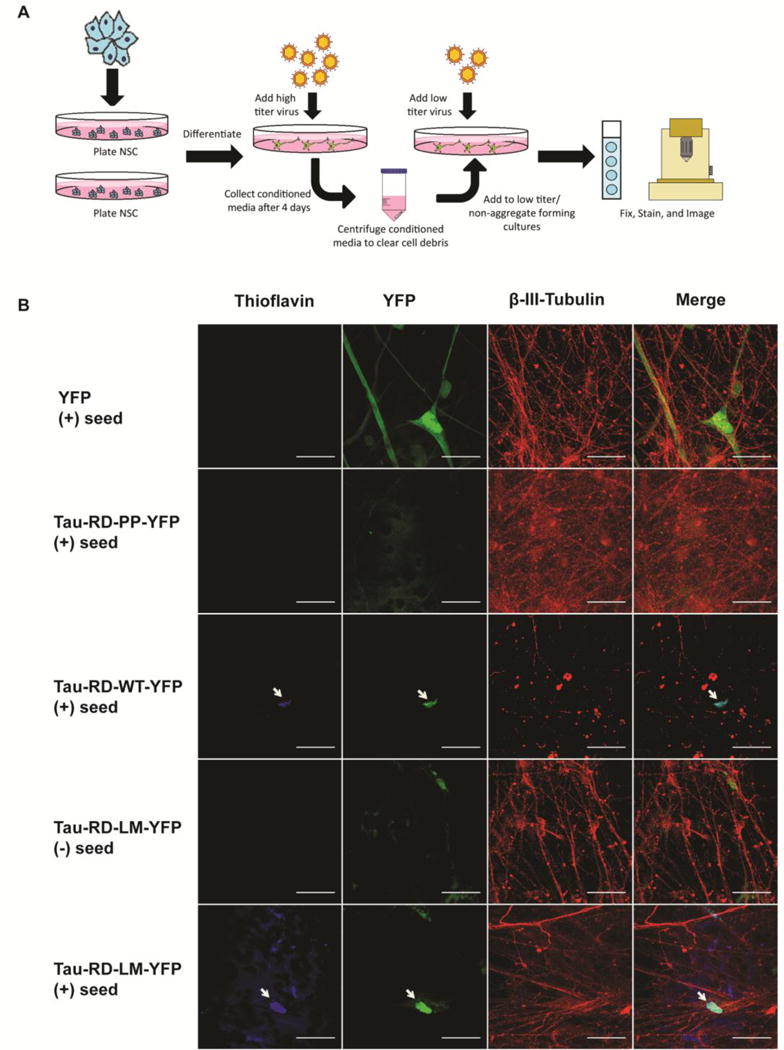
Tau-RD-LM in conditioned media mediates transcellular propagation and aggregate formation. (A) A schematic depicting the experimental set up. (B) Conditioned media was applied to human neuronal cultures minimally transduced with YFP, tau-RD-PP-YFP, tau-RD-WT-YFP or tau-RD-LM-YFP. Only cultures which were minimally transduced with tau-RD-LM-YFP, had thioflavin positive aggregates, suggesting homotypic templating. Thioflavin positive aggregates were rare but present in cultures transduced with tau-RD-WT-YFP, suggesting possible heterotypic templating. Thioflavin positive aggregates were not present in cultures expressing tau-RD-PP-YFP, indicating that the β-pleated sheet structure is required
3.5 Tau-RD induces axonal length and synaptic density loss
Synapse loss and axonal beading are early signs of neurodegeneration in AD (Selkoe, 2002). These neurodegenerative phenotypes were also observed when four-week differentiated human neuronal cultures, derived from NDC and PS-1A246E mutants were transfected with tau-RD constructs. Neurons were transfected in order to visualize them individually. Four days post-transfection, a reduction in axon length, but not dendritic length, was observed in the tau-RD constructs as compared to mCherry alone (Fig. 6A–B). In addition, a decrease in synaptic density was observed in tau-RD-LM-YFP transfected neurons (Fig. 6C). Lysosome size also increased in cells transfected with tau-RD-LM, implicating lysosomal involvement in neurodegenerative mechanisms (Fig. 6D).
Fig. 6.

Tau-RD induced reduction in axonal length and synaptic density, and enlargement of lysosomes. (A) Image of a neuron transfected with tau-RD-YFP. (B) Four-week human neuronal cultures derived from NDC or PS1 A246E mutation carrier were transfected with mCherry or with mCherry and tau-RD-WT or tau-RD-LM. Axon length is significantly reduced in neurons transfected with tau-RD-WT and tau-RD-LM in both the non-demented control (NDC) and PS1 A264E mutation carriers. (C) Dendritic length is unchanged between neurons transfected with mCherry alone or co-transfected with tau-RD-WT or tau-RD-LM. (D, E) There is a reduction in synaptic density in tau-RD transfected neurons. (F, G) Neuronal cultures transfected with tau-RD-LM show enlarged lysosomes. Lysosomal size increased with tau-RD-LM in both NDC and PS1 neurons. Lysosomal volume is expressed on x-axis as lysosomal volume/cell (μm). N = 6, biological duplicate in 3 different experiments. Statistics were performed by Student’s t test. NS = not significant, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001
3.6 NiPSCEs containing human tau injection into the mouse brain
Exosomes represent a special class of secreted vesicles that are released from most cell types and are thought to participate in waste removal and intracellular communication (Thery et al., 2002). Recent studies have characterized protein cargo and mRNAs from neuronally-derived (Fiandaca et al., 2015; Winston et al., 2016) and astrocytic-derived (Goetzl et al., 2016; Lafourcade et al., 2016) exosomes. We wanted to know whether our hiPSC system secreted exosomes into the cell culture media. We first purified exosomes in the conditioned media from NDC-derived neuronal cultures both in the presence and absence of tau-RD-LM transduction (NiPSCE-tau-RD-LM and NiPSCE, respectively). As demonstrated by nanoparticle tracking analysis (NTA), we found that the size distribution of NiPSCE fluctuated significantly while NiPSCE-tau-RD-LM was more uniform and smaller in size (Fig. 7A–C). The exosomes from the tau-RD-LM conditioned media contain human tau-RD-LM (Fig. 7D), which is recognized by K9JA, a pan-human tau antibody. K9JA does not detect human endogenous tau in the non-transduced culture media or in the non-exosomal fraction (Fig. 7D). NiPSCE-tau-RD-LM did not contain phosphorylated tau species, as recognized by PHF1 or AT8 as their epitopes are outside of the tau repeat domain (data not shown). Together, these results support that the NiPSCE-tau-RD-LM contained tau-RD-LM.
Fig. 7.

Exosomes derived from tau-RD-LM conditioned media injection in normal mouse brain. Representative NTA plot of averaged size/concentration for exosomes derived from (A) neuronal cell culture media (NiPSCE) and (B) tau-RD-LM conditioned media from induced pluripotent stem cells (NiPSCE-tau-RD-LM). (C) Averaged mean particle size results from three replicates of NiPSCE and NiPSCE-tau-RD-LM suspensions (NiPSCE, 151.7 ± 9.849 vs. NiPSCE-tau-RD-LM, 114.2 ± 0.7182 nm; Unpaired t-test * = P < 0.05). (D) Non-exosome (non-exo) and exosomal fractions were probed against various tau species. Representative western blot demonstrates that human tau (KJ9A) is only detected in the exosomal fraction derived from NiPSCE-tau-RD-LM, while the non-exo and exosomal fractions were all negative for phosphorylated tau species as recognized by PHF-1 and AT8 antibodies (data not shown). Exosome surface marker, CD63, was used as a loading control. (E) Representative photomicrographs of hippocampal sections obtained from wild-type, C57/Bl6 mice that were injected with NiPSCE-tau-RD-LM suspensions. At one-month post injection, mice injected with NiPSCE-tau-RD-LM display modest enhancement of human tau immunoreactivity in the CA1 region of the hippocampus (n = 5/group).
Next, we stereotactically injected NiPSCE-tau-RD-LM suspensions or vehicle (1x PBS) into the right hippocampus of 3–4 month-old, female, C57/Bl6 mice. One month post injection, mouse brains were sectioned and stained with K9JA to detect human tau. Human tau was visualized in the CA1 region of the hippocampus indicating that the tau-RD-LM was uptaken by hippocampal neurons (Fig. 7E). All injection control mice were negative for human tau staining (Fig. 7E).
4 Discussion
Given that abnormal levels of hyperphosphorylated tau are present in human AD brains, it is likely that tau contributes to the pathological state and may transmit pathology from cell-to-cell. Thus, it is imperative that we have a model system in place that is able to recapitulate this hallmark of neurodegeneration. The overexpression of the APP Swedish mutation and PS-1 mutation in a 3-D culture environment has been shown to cause tau phosphorylation and formation of aggregates that are SDS-insoluble and silver-stain positive (Choi et al., 2014). However, the tau lines published thus far, which include tau, presenilin, or APP mutations and duplications, as well as overexpression of full-length 4R tau, have yet to replicate NFT formation [10, 18, 20, 29, 38, 41]. This suggests that expression of mutant proteins at endogenous levels is insufficient to cause tangle formation under normal 2-D culture conditions. Here we report a novel hiPSC neuronal model of tauopathy that successfully demonstrates both early- and late-stage tau pathologies, as evidenced by neurofibrillary tangle formation. Our approach relies on expression of tau-RD, a physiologically relevant tau species that has been detected in patient cerebrospinal fluid and is a core component of pathological NFTs (Luk et al., 2012; Mori et al., 1995; Zemlan et al., 1999). Albeit tau-RD is a naturally occurring species, its aggregate-prone variant, tau-RD-LM, contains two mutations artificially expressed together. Our findings demonstrate that tau-RD is sufficient to induce aggregation, and they support the conclusion that tau propagation is related to disease progression, with tau-RD mediating cell-to-cell transfer (Frost et al., 2009; Walker et al., 2012).
Previous tau seeding experiments required incubation of recombinant fibrils and transfection of seeds or fibrils into non-human neuronal cells (Guo et al., 2013). Using our model system, tau-RD-induced NFT formation occurred without the need for exogenous seeds or fibrils. Our findings suggest that human neurons may have unique abilities to process seeds compared to non-human neuronal cells. One possibility is that there are abundant seeds in the extracellular space. We have shown that tau-RD is highly enriched in the conditioned media, which suggests that neurons and glia could be actively ejecting tau seeds into the extracellular space. It is also possible that the seeds are released into the media as a result of cell death, a hypothesis supported by the presence of tau within the CSF of AD patients (Jack et al., 2013).
It has also been shown that tau is secreted by neurons in an activity-dependent manner, similar to other pathological proteins, such as α-synuclein (Pooler et al., 2013). One related hypothesis is that tau and tau seeds are secreted from neurons as means to clear them from the cells. Understanding the mechanisms regulating this phenomenon may provide important clues regarding how seeds are cleared in the nervous system.
An alternative way to explain the spontaneous efficiency of seeding unique to human neurons is to examine the different strains of the seeds. It has been previously reported that seeds derived from human brains do not seed wild-type mouse brains (Sanders et al., 2014). There could be a species barrier making human tau distinct from that of other species, as has been demonstrated in prions (Kurt et al., 2015). It is also possible that the aggregation potential could be due to inter-species differences in tau processing. Tau aggregation formation occurs as early as young adulthood, but at a very slow rate (Braak and Del Tredici, 2011). Seed formation and propagation is likely a rare event initially, but later accelerated with aging, possibly due to a diminished proteostasis state or cross-templating from other protein aggregates (Braak and Del Tredici, 2011).
We show for the first time that tau seeds can be generated by human neurons and mediate transcellular propagation. Our data suggests homotypic aggregation is the dominant mechanism; however, heterotypic aggregation is also possible. We did not explore the possibility of cross-seeding between different proteins. Previous reports have suggested that this is possible; albeit, the specific mechanisms and parameters remain unknown (Guo et al., 2013). An exploration into cross-seeding between different proteins would be worthwhile given that many of the aggregated pathological proteins co-exist in neurodegenerative diseases; for example, tau and β-amyloid in AD, α-synuclein and β-amyloid in lewy-body dementia, and TDP-43 and tau in FTD.
Previous reports have demonstrated that prions from the prion protein PrP-Scrapie can propagate between cells via exosomes (Fevrier et al., 2004). We demonstrated that neuronal-derived exosomes are capable of transferring human tau from human neurons to neurons within naïve mouse brain. Our data supports the literature that exosomes are a modality by which tau travels between neurons.
We further observed neurodegenerative phenotypes, such as axon retraction and synapse loss. The neurotoxicity of tau-RD is independent of aggregate formation, for we observed these findings without any aggregate formation. The mechanism could be due to the toxic effect of soluble tau species. Our data would support the literature proposing soluble oligomers as the toxic species driving neurodegeneration (Lasagna-Reeves et al., 2011).
Although the human neuronal cultures at four weeks expressed mainly the immature 3R isoform, we were able to observe tau aggregation driven by tau-RD. Testing the current experimental conditions in more mature cultures expressing both 3R and 4R in the future will help to test the contribution of 4R to tau aggregation and propagation.
5. Conclusions
In conclusion, we have shown that tau seeds can be generated by human neurons and can mediate transcellular propagation. Further, we have demonstrated that tau-RD is secreted inside exosomes, which are capable of mediating the transfer of human tau into naïve mouse brains in vivo. Finally, our data suggests that human neuronal cells are more efficient at forming tau aggregates than rodent cells, suggesting potential species-dependent regulation and emphasizing the value of a more appropriate model system to study human tauopathies. Previous models using neurons derived from hiPSC of mutation carriers have demonstrated early pathologies, but failed to replicate the later-stage pathologies such as NFT formation, which our model successfully recapitulates. This human neuronal model facilitates observation of the mechanisms of tangle formation as well as the regulation of seeding and intercellular disease transmission, providing a more physiologically and pathologically relevant model for studying human tauopathies.
Supplementary Material
Highlights.
-
-
Novel human neuronal tau model exhibiting neurofibrillary tangles
-
-
Tau seeds can be generated by human neurons and can mediate transcellular propagation
-
-
Tau-repeat domain is secreted in exosomes, and can transfer into naïve mouse brains in vivo.
Acknowledgments
We thank Dr. Peter Davies for supplying antibodies (PHF1, Alz-50, CP-13), Dr. Mariko Sawa for providing rat cortical neuronal cultures, Dr. Anne Bang for critical reading of the manuscript and Stephen LeSourd for preparation of the manuscript. The Lamp2 (H4B4) antibody, developed by August and Hildreth, was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. The authors do not have conflict of interest to disclose.
Funding: This work was supported by Alzheimer’s Association [NIRG-14-322164]; California Institute for Regenerative Medicine [TB1-01193]; National Institute of Health [AG000216, AG051848, BX003040 AG0051839 and AG005131].
Abbreviations
- AD
Alzheimer’s disease
- NFT
neurofibrillary tangles
- hiPSC
human induced pluripotent stem cell
- PHF
paired helical filament
- FTD
frontotemporal dementia
- NDC
non-demented control
- PS-1
presenilin 1
- YFP
yellow fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alafuzoff I, et al. Histopathological criteria for progressive dementia disorders: clinical-pathological correlation and classification by multivariate data analysis. Acta Neuropathol. 1987;74:209–25. doi: 10.1007/BF00688184. [DOI] [PubMed] [Google Scholar]
- Alonso Adel C, et al. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279:34873–81. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, et al. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–9. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. The pathological process underlying Alzheimer, Äôs disease in individuals under thirty. Acta Neuropathologica. 2011;121:171–181. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- Choi SH, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515:274–8. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–40. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh R, et al. Lack of the carboxyl terminal sequence of tau in ghost tangles of Alzheimer’s disease. Brain Res. 1993;601:164–72. doi: 10.1016/0006-8993(93)91707-y. [DOI] [PubMed] [Google Scholar]
- Fevrier B, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101:9683–8. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiandaca MS, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015;11:600–7 e1. doi: 10.1016/j.jalz.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong H, et al. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Reports. 2013;1:226–34. doi: 10.1016/j.stemcr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, et al. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–52. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallyas F. Silver staining of Alzheimer’s neurofibrillary changes by means of physical development. Acta Morphol Acad Sci Hung. 1971;19:1–8. [PubMed] [Google Scholar]
- Garcia-Sierra F, et al. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer’s disease. J Alzheimers Dis. 2003;5:65–77. doi: 10.3233/jad-2003-5201. [DOI] [PubMed] [Google Scholar]
- Goetzl EJ, et al. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016;30:3853–3859. doi: 10.1096/fj.201600756R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–17. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, et al. Transgenic Mouse Model of Tauopathies with Glial Pathology and Nervous System Degeneration. Neuron. 2002;35:433–446. doi: 10.1016/s0896-6273(02)00789-4. [DOI] [PubMed] [Google Scholar]
- Holmes BB, et al. Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci U S A. 2014;111:E4376–85. doi: 10.1073/pnas.1411649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Iovino M, et al. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain. 2015;138:3345–59. doi: 10.1093/brain/awv222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, et al. Mechanisms of tau-induced neurodegeneration. Acta Neuropathologica. 2009;118:53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–20. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha GA, et al. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–32. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Kfoury N, et al. Trans-cellular Propagation of Tau Aggregation by Fibrillar Species. J Biol Chem. 2012;287:19440–51. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatoon S, et al. Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem. 1992;59:750–3. doi: 10.1111/j.1471-4159.1992.tb09432.x. [DOI] [PubMed] [Google Scholar]
- Khatoon S, et al. Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett. 1994;351:80–4. doi: 10.1016/0014-5793(94)00829-9. [DOI] [PubMed] [Google Scholar]
- Khlistunova I, et al. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–14. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- Khlistunova I, et al. Inhibition of tau aggregation in cell models of tauopathy. Curr Alzheimer Res. 2007;4:544–6. doi: 10.2174/156720507783018307. [DOI] [PubMed] [Google Scholar]
- Knopman DS, et al. Brain injury biomarkers are not dependent on β -amyloid in normal elderly. Annals of Neurology. 2013;73:472–480. doi: 10.1002/ana.23816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt TD, et al. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. The Journal of Clinical Investigation. 2015;125:1485–1496. doi: 10.1172/JCI79408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafourcade C, et al. MiRNAs in Astrocyte-Derived Exosomes as Possible Mediators of Neuronal Plasticity. J Exp Neurosci. 2016;10:1–9. doi: 10.4137/JEN.S39916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, et al. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, et al. Effect of potent gamma-secretase modulator in human neurons derived from multiple presenilin 1-induced pluripotent stem cell mutant carriers. JAMA Neurol. 2014;71:1481–9. doi: 10.1001/jamaneurol.2014.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk C, et al. Development and assessment of sensitive immuno-PCR assays for the quantification of cerebrospinal fluid three- and four-repeat tau isoforms in tauopathies. J Neurochem. 2012;123:396–405. doi: 10.1111/j.1471-4159.2012.07911.x. [DOI] [PubMed] [Google Scholar]
- Mandelkow E, et al. Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol. 2007;17:83–90. doi: 10.1111/j.1750-3639.2007.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, et al. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer’s disease. J Neurosci. 1996;16:5795–811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medda X, et al. Development of a Scalable, High-Throughput-Compatible Assay to Detect Tau Aggregates Using iPSC-Derived Cortical Neurons Maintained in a Three-Dimensional Culture Format. J Biomol Screen. 2016;21:804–15. doi: 10.1177/1087057116638029. [DOI] [PubMed] [Google Scholar]
- Mertens J, et al. Embryonic Stem Cell,ÄìBased Modeling of Tau Pathology in Human Neurons. The American Journal of Pathology. 2013;182:1769–1779. doi: 10.1016/j.ajpath.2013.01.043. [DOI] [PubMed] [Google Scholar]
- Mitsuhashi M, et al. Aging enhances release of exosomal cytokine mRNAs by Abeta-1-42-stimulated macrophages. FASEB J. 2013;27:5141–50. doi: 10.1096/fj.13-238980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragon-Rodriguez S, et al. Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer’s disease. Int J Exp Pathol. 2008;89:81–90. doi: 10.1111/j.1365-2613.2007.00568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, et al. Tau in cerebrospinal fluids: establishment of the sandwich ELISA with antibody specific to the repeat sequence in tau. Neurosci Lett. 1995;186:181–3. doi: 10.1016/0304-3940(95)11291-4. [DOI] [PubMed] [Google Scholar]
- Novak M, et al. Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci U S A. 1991;88:5837–41. doi: 10.1073/pnas.88.13.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, et al. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–94. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, et al. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172–7. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–83. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman RA, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–30. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–88. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Thery C, et al. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–79. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- Valadi H, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- Verheyen A, et al. Using Human iPSC-Derived Neurons to Model TAU Aggregation. PLoS One. 2015;10:e0146127. doi: 10.1371/journal.pone.0146127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, et al. Mechanisms of Protein Seeding in Neurodegenerative Diseases. Arch Neurol. 2012:1–7. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston CN, et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement (Amst) 2016;3:63–72. doi: 10.1016/j.dadm.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SH, et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 2011;6:e17540. doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SH, Shaner M. Bioengineered stem cells in neural development and neurodegeneration research. Ageing Res Rev. 2013;12:739–48. doi: 10.1016/j.arr.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemlan FP, et al. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem. 1999;72:741–50. doi: 10.1046/j.1471-4159.1999.0720741.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.